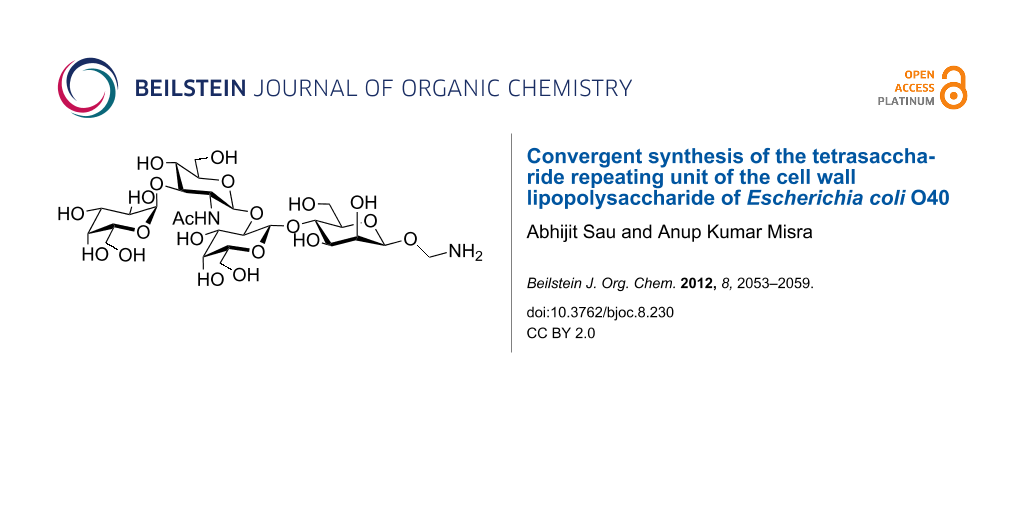
Abstract
A tetrasaccharide repeating unit corresponding to the cell-wall lipopolysaccharide of E. coli O40 was synthesized by using a convergent block glycosylation strategy. A disaccharide donor was coupled to a disaccharide acceptor by a stereoselective glycosylation. A 2-aminoethyl linker was chosen as the anomeric protecting group at the reducing end of the tetrasaccharide. All glycosylation steps are significantly high yielding and stereoselective.

Graphical Abstract
Introduction
Infantile diarrhoea is one of the major causes of morbidity and mortality in infancy in developing countries [1]. Among several factors, Escherichia coli (E. coli) infection is one of the major causes of diarrhoeal disease in the developing countries [2]. E. coli are Gram-negative opportunistic pathogens and belong to the genus Enterobacteriaceae. In general, E. coli is considered as a friendly organism present in the normal intestinal flora of humans and animals and can kill harmful bacteria by producing vitamins and other immunostimulants [3]. However, a number of E. coli strains acquire virulence factors and cause severe intestinal and urinary-tract infections [4,5]. E. coli serotypes are generally classified based on the somatic, flagella and capsular antigens [6]. Diarrhoea-causing E. coli strains are broadly classified in four categories: (a) Enteropathogenic E. coli infects through the production of heat-labile and heat-stable toxins; (b) enteroinvasive E. coli acts through the invasion of the host body; (c) enteropathogenic E. coli infects by adhering to the membrane of the host intestine; and (d) verotoxin E. coli infects by the production of verotoxin or shiga toxin [7]. Recently, Zhao et al. reported the structure of the repeating unit of the cell-wall antigenic lipopolysaccharide of E. coli O40 [8], which contains two D-galactosyl moieties with alpha and beta linkage, one beta-linked D-glucosamine and one beta-linked D-mannosyl moiety (Figure 1).
![[1860-5397-8-230-1]](/bjoc/content/figures/1860-5397-8-230-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the tetrasaccharide repeating unit of the cell-wall lipopolysaccharide of Escherichia coli O40.
Figure 1: Structure of the tetrasaccharide repeating unit of the cell-wall lipopolysaccharide of Escherichia ...
Although several therapeutics have appeared in the past to control the diarrheal epidemics caused by E. coli infections, emergence of resistant strains is a serious concern in the development of therapeutics against this organism. Since, bacterial cell-wall lipopolysaccharides play important roles in the pathogenicity of the virulent strains, it would be pertinent to develop glycoconjugate therapeutics based on the cell-wall oligosaccharide haptens to reduce the number of infections [9-12]. In order to evaluate the therapeutic efficacy of the glycoconjugate derivatives it is essential to have a significant quantity of oligosaccharides, which is difficult to isolate from natural sources. Therefore, the development of a chemical synthetic strategy for the synthesis of the oligosaccharides and their close analogues can add momentum towards the preparation of glycoconjugate-based therapeutics. In this perspective, we report herein a concise chemical synthesis of the tetrasaccharide repeating unit of the cell-wall lipopolysaccharide of E. coli O40, using a convergent block synthetic strategy.
Results and Discussion
The target tetrasaccharide 1 as its 2-aminoethyl glycoside was synthesized by a stereoselective glycosylation of a disaccharide acceptor 8 and a disaccharide thioglycoside donor 9 using a [2 + 2] block synthetic strategy. The disaccharide intermediates were synthesized from the suitably protected monosaccharide derivatives 2 [13], 3 [14], 4 [15] and 5 [16], which were prepared from the commercially available reducing sugars, by applying a series of functional group protection–deprotection methodologies (Figure 2). The synthetic strategy has a number of notable features, which include (a) stereoselective [2 + 2] block glycosylation; (b) application of general glycosylation reactions by using thioglycosides as glycosyl donors and a combination of N-iodosuccinimide (NIS) and perchloric acid supported over silica (HClO4–SiO2) [17,18] as glycosyl activator; (c) exploitation of the armed–disarmed glycosylation concept for the orthogonal activation of thioglycoside during the synthesis of disaccharide derivative 9 [19]; (d) use of aminoethyl linker as the anomeric protecting group; (e) removal of benzyl groups using a combination of triethylsilane and Pd(OH)2–C [20]; and (f) preparation of β-D-mannosidic moiety from the β-D-glucoside [13].
![[1860-5397-8-230-2]](/bjoc/content/figures/1860-5397-8-230-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of the synthesized tetrasaccharide 1 and its synthetic precursors.
Figure 2: Structure of the synthesized tetrasaccharide 1 and its synthetic precursors.
Benzylation of 2-azidoethyl 3-O-benzyl-4,6-O-benzylidene-β-D-mannopyranoside (2) [13] (prepared from D-glucose in nine steps) by using benzyl bromide and sodium hydroxide [21] followed by reductive ring opening of the 4,6-O-benzylidene acetal with triethylsilane and iodine [22] furnished compound 6 in 82% yield. Stereoselective glycosylation of compound 6 with thioglycoside derivative 3 in the presence of a combination of N-iodosuccinimide (NIS) and HClO4–SiO2 [17] gave disaccharide derivative 7 in a 77% yield. Formation of compound 7 was confirmed from its spectral analysis [signals at δ 4.73 (d, J = 8.0 Hz, H-1B), 4.41 (br s, H-1A) in the 1H NMR and at δ 101.6 (C-1A), 100.7 (C-1B) in the 13C NMR spectra respectively]. Saponification of compound 7 by using sodium methoxide followed by 3,4-O-isopropylidenation with 2,2-dimethoxypropane and p-toluenesulfonic acid [23] furnished disaccharide derivative 8 in 74% yield (Scheme 1).
![[1860-5397-8-230-i1]](/bjoc/content/inline/1860-5397-8-230-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of disaccharide derivative 8. Reagents and conditions: (a) benzyl bromide, NaOH, DMF, room temperature, 1 h; (b) Et3SiH, I2, CH3CN, 0 °C, 1 h, 82%; (c) NIS, HClO4–SiO2, MS 4Å, CH2Cl2, −25 °C, 1 h, 77%; (d) 0.1 M CH3ONa, CH3OH, room temperature, 3 h; (e) 2,2-dimethoxypropane, p-TsOH, DMF, room temperature, 5 h, 74%.
Scheme 1: Synthesis of disaccharide derivative 8. Reagents and conditions: (a) benzyl bromide, NaOH, DMF, roo...
In a separate experiment, stereoselective glycosylation of thioglycoside derivative 4 with the thioglycoside acceptor 5 in the presence of a combination of NIS and HClO4–SiO2 [17] in dichloromethane–diethyl ether furnished disaccharide thioglycoside derivative 9 in a 74% yield together with a minor quantity of its other isomer (≈5%), which was separated by column chromatography. Formation of compound 9 was confirmed from its spectral analysis [δ 5.51 (d, J = 3.5 Hz, H-1D), 5.37 (d, J = 10.5 Hz, H-1C) in the 1H NMR and δ 97.4 (C-1D), 83.0 (C-1C) in the 13C NMR spectra, respectively]. During the synthesis of compound 9, thioglycoside 4 acted as glycosyl donor and thioglycoside 5 acted as orthogonal glycosyl acceptor because of the difference in their reactivity following the “armed–disarmed glycosylation” concept [19,24] (Scheme 2).
![[1860-5397-8-230-i2]](/bjoc/content/inline/1860-5397-8-230-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of disaccharide derivative 9. Reagents and conditions: (a) NIS, HClO4–SiO2, MS 4Å, CH2Cl2–Et2O, −25 °C, 1 h, 74%.
Scheme 2: Synthesis of disaccharide derivative 9. Reagents and conditions: (a) NIS, HClO4–SiO2, MS 4Å, CH2Cl2...
Iodonium ion promoted [2 + 2] stereoselective glycosylation of compound 8 and compound 9 in the presence of NIS and HClO4–SiO2 [17] furnished tetrasaccharide derivative 10 in 71% yield. Formation of compound 10 was confirmed by its spectral analysis [signals at δ 101.6 (C-1B), 101.5 (PhCH), 100.8 (C-1C), 100.2 (C-1A), 97.3 (C-1D) in the 13C NMR spectrum]. Compound 10 was subjected to a sequence of reactions involving (a) removal of N-phthalimido group by using hydrazine hydrate [25]; (b) N-acetylation by using acetic anhydride and pyridine; (c) removal of isopropylidene ketal and benzylidene acetal by acid hydrolysis; and finally (d) removal of benzyl ethers by using triethylsilane and 20% Pd(OH)2–C [20] to furnish target compound 1, which was purified through a Sephadex® LH-20 column to give pure compound 1 in 60% overall yield. Spectral data of compound 1 confirmed its formation [signals at δ 5.31 (d, J = 8.5 Hz, H-1C), 5.15 (d, J = 3.5 Hz, H-1D), 4.63 (br s, H-1A), 4.34 (d, J = 8.5 Hz, H-1B) in the 1H NMR and at δ 100.7 (C-1B), 100.6 (C-1B), 99.6 (2 C, C-1A, C-1C) in the 13C NMR] (Scheme 3).
![[1860-5397-8-230-i3]](/bjoc/content/inline/1860-5397-8-230-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of target tetrasaccharide 1. Reagents and conditions: (a) NIS, HClO4–SiO2, CH2Cl2, −15 °C, 1 h, 71%; (b) NH2NH2·H2O, EtOH, 90 °C, 5 h; (c) acetic anhydride, pyridine, room temperature, 2 h; (d) 80% aq AcOH, 80 °C, 1.5 h; (e) triethylsilane, 20% Pd(OH)2–C, CH3OH, 6 h, room temperature, overall 60%.
Scheme 3: Synthesis of target tetrasaccharide 1. Reagents and conditions: (a) NIS, HClO4–SiO2, CH2Cl2, −15 °C...
Conclusion
In summary, synthesis of a tetrasaccharide repeating unit corresponding to the cell-wall lipopolysaccharide of E. coli O40 was achieved by using a convergent [2 + 2] block synthetic strategy. The yields are excellent in all reactions. A general reaction condition was used in all glycosylation reactions. All intermediates and final compounds were characterized by their spectral analysis. The armed–disarmed glycosylation concept was applied for the synthesis of disaccharide derivative 9. A 2-Aminoethyl linker was used as the anomeric protecting group.
Experimental
General methods: All reactions were monitored by thin-layer chromatography over silica-gel-coated TLC plates. The spots on TLC were visualized by warming ceric sulfate (2% Ce(SO4)2 in 2 N H2SO4)-sprayed plates on a hot plate. Silica gel 230–400 mesh was used for column chromatography. 1H and 13C NMR spectra were recorded on Brucker Avance 500 MHz by using CDCl3 as solvent and TMS as internal reference, unless stated otherwise. Chemical shift values are expressed in δ ppm. MALDI-MS were recorded on a Bruker Daltronics mass spectrometer. Commercially available grades of organic solvents of adequate purity were used in all reactions. HClO4–SiO2 was prepared following the method reported in the literature [18].
2-Azidoethyl 2,3,6-tri-O-benzyl-β-D-mannopyranoside (6): To a solution of compound 2 (2.0 g, 4.68 mmol) in dry DMF (10 mL) were added benzyl bromide (1.2 mL, 10.09 mmol) and powdered NaOH (750.0 mg, 18.75 mmol) and the reaction mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with water (100 mL) and extracted with CH2Cl2 (100 mL). The organic layer was washed with H2O, dried (Na2SO4) and concentrated. The crude product was passed through a short pad of SiO2 by using hexane–EtOAc (5:1) as eluant to give the O-benzylated product (2.2 g, 91%). A solution of the O-benzylated product (2.2 g, 4.25 mmol) in dry CH3CN (20 mL) was cooled to 0 °C. To the cooled reaction mixture were added Et3SiH (1.4 mL, 8.76 mmol) and I2 (250.0 mg, 0.98 mmol), and the reaction mixture was stirred at the same temperature for 1 h. The reaction mixture was diluted with CH2Cl2 (100 mL) and the organic layer was successively washed with saturated NaHCO3 and H2O, and then dried (Na2SO4) and concentrated. The crude product was purified over SiO2 by using hexane–EtOAc (4:1) as eluant to give pure compound 6 (1.7 g, overall 82%). White solid; mp 89–90 °C; [α]D25 −97 (c 1.0, CHCl3); IR (KBr): 3293, 2845, 2110, 1497, 1454, 1365, 1310, 1119, 1065, 779, 659, 599 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.43–7.23 (m, 15H, Ar-H), 4.98 (d, J = 12.5 Hz, 1H, PhCH2), 4.75 (d, J = 12.5 Hz, 1H, PhCH2), 4.61, 4.58 (2 d, J = 12.0 Hz, 2H, PhCH2), 4.48 (br s, 1H, H-1), 4.45 (d, J = 12.0 Hz, 1H, PhCH2), 4.32 (d, J = 12.0 Hz, 1H, PhCH2), 4.15–4.11 (m, 1H,-OCH2-), 3.96 (br s, 1H, H-2), 3.94 (t, J = 9.5 Hz each, 1H, H-4), 3.85 (dd, J = 10.5, 3.5 Hz, 1H, H-6a), 3.75 (dd, J = 10.5, 6.5 Hz, 1H, H-6b), 3.66–3.62 (m, 1H, OCH2-), 3.58–3.53 (m, 1H, CH2N3), 3.46–3.42 (m, 1H, H-5), 3.34–3.30 (m, 1H, CH2N3), 3.29 (dd, J = 10.0, 3.5 Hz, 1H, H-3); 13C NMR (125 MHz, CDCl3) δ 138.6–127.4 (Ar-C), 101.8 (C-1), 81.3 (C-3), 75.4 (C-5), 74.4 (PhCH2), 73.7 (PhCH2), 73.5 (C-5), 71.1 (PhCH2), 70.7 (C-6), 68.6 (OCH2), 68.0 (C-2), 50.9 (CH2N3); ESI-MS: 542.2 [M + Na]+; Anal. calcd for C29H33N3O6: C, 67.04; H, 6.40; found: C, 66.90; H, 6.58.
2-Azidoethyl O-(2,3,4-tri-O-acetyl-6-O-benzyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-mannopyranoside (7): To a solution of compound 3 (1.4 g, 3.18 mmol) and compound 6 (1.5 g, 2.88 mmol) in anhydrous CH2Cl2 (10 mL) was added MS 4Å (2.0 g), and the reaction mixture was stirred at room temperature for 30 min under argon. The reaction mixture was cooled to −25 °C, and N-iodosuccinimide (NIS; 0.8 g, 3.55 mmol) and HClO4–SiO2 (25.0 mg) were added to it. After being stirred at same temperature for 1 h the reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 mL). The organic layer was successively washed with 5% Na2S2O3, saturated NaHCO3 and water, and then dried (Na2SO4) and concentrated under reduced pressure to give the crude product. The crude product was purified over SiO2 by using hexane–EtOAc (7:1) as eluant to give pure compound 7 (2.0 g, 77%). Yellow oil; [α]D25 −13 (c 1.0, CHCl3); IR (neat): 3087, 2956, 2153, 1605, 1487, 1345, 1254, 1183, 1142, 1045, 999, 774, 734, 647, 542 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.35–7.16 (m, 20H, Ar-H), 5.33 (d, J = 3.0 Hz, 1H, H-4B), 5.06 (t, J = 8.0 Hz each, 1H, H-2B), 4.87 (d, J = 12.0 Hz, 1H, PhCH2), 4.84 (dd, J = 10.5, 3.5 Hz, 1H, H-3B), 4.73 (d, J = 8.0 Hz, 1H, H-1B), 4.72–4.71 (2 d, J = 12.0 Hz each, 2H, PhCH2), 4.59 (d, J = 12.0 Hz, 1H, PhCH2), 4.47, 4.45 (2 d, J = 12.0 Hz each, 2H, PhCH2), 4.41 (br s, 1H, H-1A), 4.39 (d, J = 12.0 Hz, 1H, PhCH2), 4.19 (d, J = 12.0 Hz, 1H, PhCH2), 4.14 (t, J = 9.0 Hz each, 1H, H-4A), 4.10–4.05 (m, 1H, OCH2), 3.92 (d, J = 3.0 Hz, 1H, H-2A), 3.76–3.70 (m, 2H, H-6abA), 3.65–3.60 (m, 1H, OCH2), 3.54–3.49 (m, 2H, H-5B, CH2N3), 3.47 (dd, J = 10.0, 3.0 Hz, 1H, H-3A), 3.42–3.38 (m, 1H, H-5A), 3.32–3.22 (m, 3H, H-6abB, CH2N3), 1.99, 1.95, 1.90 (3 s, 9H, 3 COCH3); 13C NMR (125 MHz, CDCl3) δ 169.9, 169.8, 169.4 (3 COCH3), 138.5–126.8 (Ar-C), 101.6 (C-1A), 100.7 (C-1B), 80.6 (C-5B), 75.7 (C-5A), 74.7 (C-2A), 74.3 (2 C, C-4A, PhCH2), 73.7 (PhCH2), 73.3 (PhCH2), 71.7 (C-5B), 71.5 (PhCH2), 71.3 (C-3B), 70.0 (C-2B), 68.6 (C-6A), 68.5 (OCH2), 67.4 (C-4B), 66.9 (C-6B), 50.8 (CH2N3), 20.7, 20.6, 20.5 (COCH3); MALDI-MS: 920.3 [M + Na]+; Anal. calcd for C48H55N3O14: C, 64.20; H, 6.17; found: C, 64.06; H, 6.35.
2-Azidoethyl O-(6-O-benzyl-3,4-O-isopropylidene-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-mannopyranoside (8): A solution of compound 7 (1.8 g, 2.0 mmol) in 0.1 M CH3ONa (25 mL) was stirred at room temperature for 2 h. The reaction mixture was neutralized with Dowex 50W X8 (H+) resin, filtered and concentrated. To a solution of the de-O-acetylated product in dry DMF (10 mL) was added 2,2-dimethoxypropane (0.7 mL, 5.69 mmol) followed by p-TsOH (0.2 g) and the reaction mixture was stirred at room temperature for 5 h. The reaction was quenched with Et3N (1 mL), the solvents were removed under reduced pressure, and the crude reaction mixture was diluted with CH2Cl2 (100 mL). The organic layer was washed with saturated NaHCO3, dried (Na2SO4) and concentrated to give the crude product, which was purified over SiO2 by using hexane–EtOAc (2:1) as eluant to give pure compound 8 (1.2 g, 74%). Yellow oil; [α]D25 −21 (c 1.0, CHCl3); IR (neat): 3418, 3030, 2926, 2198, 1743, 1711, 1646, 1390, 1253, 1099, 1053, 864, 754, 667, 531 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.30–7.14 (m, 20H, Ar-H), 4.84 (d, J = 12.5 Hz, 1H, PhCH2), 4.63–4.40 (m, 6H, PhCH2), 4.38 (d, J = 8.0 Hz, 1H, H-1B), 4.33 (br s, 1H, H-1A), 4.30 (d, J = 12.5 Hz, 1H, PhCH2), 4.22 (t, J = 9.5 Hz each, 1H, H-4A), 4.02–3.98 (m, 2H, H-2A, OCH2), 3.91 (dd, J = 10.0, 3.5 Hz, H-3B), 3.83 (dd, J = 12.0, 5.5 Hz, 1H, H-6aB), 3.81 (d, J = 2.0 Hz, 1H, H-4B), 3.76 (dd, J = 12.0, 2.0, Hz, 1H, H-6bB), 3.71 (br s, 1H, H-5B), 3.70–3.68 (m, 1H, OCH2), 3.62–3.58 (m, 1H, H-6aA), 3.56–3.51 (m, 1H, CH2N3), 3.49 (dd, J = 8.0 Hz each, 1H, H-2B), 3.47–3.43 (m, 1H, H-6bA), 3.42–3.37 (m, 2H, H-3A, H-5A), 3.25–3.19 (m, 1H, CH2N3), 1.42, 1.25 (2 s, 6H, 2 C(CH3)3); 13C NMR (125 MHz, CDCl3) δ 138.7–127.3 (Ar-C), 109.8 (C(CH3)2), 102.5 (C-1B), 101.9 (C-1A), 80.6 (C-2B), 78.9 (C-3B), 75.2 (C-5A), 74.3 (PhCH2), 74.2 (C-3A), 74.0 (C-4B), 73.8 (C-2A), 73.5 (PhCH2), 73.4 (2C, C-4A, PhCH2), 72.4 (C-5B), 71.3 (PhCH2), 69.4 (OCH2), 69.3 (C-6B), 68.5 (C-6A), 50.8 (CH2N3), 28.2, 26.4 (C(CH3)2); MALDI-MS: 834.3 [M + Na]+; Anal. calcd for C45H53N3O11: C, 66.57; H, 6.58; found: C, 66.42; H, 6.75.
Ethyl O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-(1→3)-4,6-O-benzylidene-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (9): To a solution of compound 4 (1.4 g, 2.39 mmol) and compound 5 (1.0 g, 2.26 mmol) in anhydrous CH2Cl2–Et2O (10 mL; 1:1 v/v) was added MS 4Å (2.0 g), and reaction mixture was stirred at room temperature for 30 min under argon. The reaction mixture was cooled to −25 °C and NIS (550.0 mg, 2.44 mmol) and HClO4–SiO2 (15.0 mg) were added. After being stirred at same temperature for 1 h the reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 mL). The organic layer was successively washed with 5% Na2S2O3, saturated NaHCO3 and water, and then dried (Na2SO4) and concentrated under reduced pressure to give the crude product. The crude product was purified over SiO2 by using hexane–EtOAc (7:1) as eluant to give pure compound 9 (1.6 g, 74%). White solid; mp 67–68 °C; [α]D25 +39 (c 1.0, CHCl3); IR (KBr): 3417, 3063, 2870, 1774, 1715, 1610, 1495, 1485, 1385, 1216, 1099, 1023, 914, 753, 719 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.76–6.91 (m, 29H, Ar-H), 5.51 (d, J = 3.5 Hz, 1H, H-1D), 5.37 (d, J = 10.5 Hz, 1H, H-1C), 5.32 (s, 1H, PhCH), 4.84 (t, J = 9.5 Hz each, 1H, H-3C), 4.77–4.58 (3 d, J = 12.0 Hz each, 3H, PhCH2), 4.46 (t, J = 9.5 Hz each, 1H, H-2C), 4.44 (d, J = 11.5 Hz, 1H, PhCH2), 4.34 (d, J = 11.5 Hz, 1H, PhCH2), 4.29 (t, J = 9.5 Hz each, 1H, H-4C), 4.19 (d, J = 11.5 Hz, 1H, PhCH2), 3.86 (br s, 2H, PhCH2), 3.85 (br s, 1H, H-4D), 3.81 (dd, J = 10.5, 3.0 Hz, 1H, H-2D), 3.72–3.67 (m, 3H, H-3D, H-5C, H-6aD), 3.58 (br s, 1H, H-5D), 3.33–3.31 (m, 1H, H-6bD), 3.23–3.19 (m, 1H, H-6aC), 2.80–2.77 (m, 1H, H-6bC), 2.67–2.56 (m, 2H, SCH2CH3), 1.12 (t, J = 7.5 Hz each, 3H, SCH2CH3); 13C NMR (125 MHz, CDCl3) δ 168.1, 167.9 (PhthCO), 138.9–123.1 (Ar-C), 101.7 (PhCH), 97.4 (C-1D), 83.0 (C-1C), 81.7 (C-4D), 78.1 (C-3D), 75.4 (C-2D), 74.8 (C-5D), 74.7 (PhCH2), 73.3 (2 C, C-3C, PhCH2), 72.8 (PhCH2), 71.8 (PhCH2), 70.1 (C-5C), 69.4 (C-4C), 68.8 (C-6D), 67.7 (C-6C), 54.2 (C-2C), 24.0 (SCH2CH3), 14.9 (SCH2CH3); MALDI-MS: 986.3 [M + Na]+; Anal. calcd for C57H57NO11S: C, 71.01; H, 5.96; found: C, 70.88; H, 6.13.
2-Azidoethyl O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-(1→3)-O-(4,6-O-benzylidene-2-deoxy-2-N-phthalimido-β-D-glucopyranosyl)-(1→2)-O-(6-O-benzyl-3,4-O-isopropylidene-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-mannopyranoside (10): To a solution of compound 8 (1.0 g, 1.23 mmol) and compound 9 (1.3 g, 1.35 mmol) in anhydrous CH2Cl2 (10 mL) was added MS 4Å (2.0 g), and reaction mixture was stirred at room temperature for 30 min under argon. The reaction mixture was cooled to −25 °C and NIS (350.0 mg, 1.55 mmol) and HClO4–SiO2 (10.0 mg) were added to it. After being stirred at same temperature for 1 h the reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 mL). The organic layer was successively washed with 5% Na2S2O3, saturated NaHCO3 and water, and then dried (Na2SO4) and concentrated under reduced pressure to give the crude product. The crude product was purified over SiO2 by using hexane–EtOAc (7:1) as eluant to give pure compound 10 (1.5 g, 71%). White solid; mp 65–66 °C; [α]D25 +34 (c 1.0, CHCl3); IR (KBr): 3423, 3063, 3030, 2871, 2105, 1776, 1744, 1715, 1497, 1454, 1389, 1239, 1102, 1060, 874, 737, 721, 697, 600, 530 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.76–6.96 (m, 49H, Ar-H), 5.52 (d, J = 3.5 Hz, 1H, H-1D), 5.42 (d, J = 8.0 Hz, 1H, H-1C), 5.25 (s, 1H, PhCH), 4.86–4.45 (m, 13H, PhCH2), 4.41 (d, J = 9.5 Hz, 1H, H-1B), 4.37 (t, J = 8.0 Hz, 1H, H-2C), 4.29 (d, J = 11.5 Hz, 1H, PhCH2), 4.24–4.20 (m, 4H, H-1A, H-3C, H-4A, OCH2), 4.13–4.08 (m, 2H, H-2A, OCH2), 3.91 (br s, 2H, PhCH2), 3.89–3.85 (m, 3H, H-2D, H-3B, H-3D), 3.84–3.75 (m, 3H, H-2B, H-4C, H-4D), 3.70 (t, J = 10.5 Hz each, 1H, H-6aB), 3.64–3.60 (m, 4H, H-4B, H-5B, H-5D, H-6bB), 3.58–3.43 (m, 5H, H-3A, H-6aA, H-6aC, H-6abD), 3.42–3.36 (m, 4H, H-5A, H-6bA, H-6bC, CH2N3), 3.34–3.26 (m, 2H, H-5C, CH2N3), 1.27, 1.25 (2 s, 6H, 2 CH3); 13C NMR (125 MHz, CDCl3) δ 138.6–126.3 (Ar-C), 109.5 (C(CH3)2), 101.6 (C-1B), 101.5 (PhCH), 100.8 (C-1C), 100.2 (C-1A), 97.3 (C-1D), 82.9 (C-4C), 82.7 (C-5A), 80.1 (C-5c), 78.7 (C-3A), 78.1 (C-4D), 76.2 (C-2D), 75.5 (C-2B), 74.9 (C-3B), 74.8 (C-5D), 74.7 (PhCH2), 74.6 (C-3D), 73.9 (PhCH2) 73.5 (C-3C), 73.4 (2 C, 2 PhCH2), 73.2 (PhCH2), 72.8 (PhCH2), 72.6 (C-2A), 72.0 (PhCH2), 71.7 (2 C, C-4B, PhCH2), 69.3 (C-4A), 68.9 (C-6C), 68.8 (OCH2), 68.7 (C-6A), 68.4 (OCH2), 67.7 (2 C, C-6B, C-6D), 65.4 (C-5B), 56.0 (C-2C), 50.8 (CH2N3), 27.6, 25.7 (C(CH3)2); MALDI-MS: 1735.7 [M + Na]+; Anal. calcd for C100H104N4O22: C, 70.08; H, 6.12; found: C, 69.94; H, 6.30.
2-Aminoethyl (α-D-galactopyranosyl)-(1→3)-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1→2)-(β-D-galactopyranosyl)-(1→4)-β-D-mannopyranoside (1): To a solution of compound 10 (500.0 mg, 0.29 mmol) in EtOH (5 mL) was added NH2NH2·H2O (0.1 mL) and the reaction mixture was stirred at 90 °C for 5 h. The solvents were removed under reduced pressure, and a solution of the crude product in acetic anhydride–pyridine (2 mL, 1:1 v/v) was kept at room temperature for 2 h and then concentrated. A solution of the crude product in 80% aq AcOH (10 mL) was stirred at 80 °C for 1.5 h and then concentrated. To a solution of the crude product in CH3OH (5 mL) were added Et3SiH (1.5 mL, 9.39 mmol) and 20% Pd(OH)2–C (100.0 mg) and the reaction mixture was stirred at room temperature for 6 h. The reaction mixture was filtered through a Celite® bed and washed with CH3OH–H2O (2:1). The solvents were removed under reduced pressure and the product was passed through a Sephadex® LH-20 column by using CH3OH–H2O (3:1) as eluant to furnish pure compound 1 (135.0 mg, 60%). Glass; [α]D25 +29 (c 1.0, H2O); IR (KBr): 3436, 2948, 1619, 1369, 1162, 669 cm−1; 1H NMR (500 MHz, D2O) δ 5.31 (d, J = 8.5 Hz, 1H, H-1C), 5.15 (d, J = 3.5 Hz, 1H, H-1D), 4.63 (br s, 1H, H-1A), 4.48 (t, J = 10.5 Hz each, 1H, H-3C), 4.34 (d, J = 8.5 Hz, 1H, H-1B), 4.09 (t, J = 10.0 Hz each, 1H, H-2C), 4.05–3.92 (m, 4H, H-2A, H-4D, H-5D, OCH2a), 3.90–3.80 (m, 4H, H-2B, H-3A, H-6aB, OCH2b), 3.78–3.57 (m, 11H, H-3D, H-4B, H-4C, H-6abA, H-6bB, H-6abC, H-6abD), 3.55–3.47 (m, 2H, H-2D, H-3B), 3.45–3.40 (m, 2H, H-5A, H-5B), 3.35–3.33 (m, 1H, H-5C), 3.20–3.15 (m, 2H, CH2NH2), 2.06 (s, 3H, COCH3); 13C NMR (125 MHz, D2O) δ 171.5 (COCH3), 100.7 (C-1B), 100.6 (C-1B), 99.6 (2C, C-1A, C-1C), 81.7 (C-3B), 79.0 (C-3C), 78.5 (C-4D), 77.0 (C-3D), 75.5 (C-4A), 75.1 (C-4B), 73.5 (C-2D), 73.3 (C-5A), 71.3 (C-2A), 70.7 (C-4C), 70.6 (C-3A), 69.8 (2 C, C-5C, C-5D), 68.3 (2C, C-2B, C-5B), 65.5 (OCH2), 60.6 (C-6B), 60.5 (C-6C), 60.2 (C-6A), 59.2 (C-6D), 55.7 (C-2C), 39.5 (CH2NH2), 23.1 (COCH3); MALDI-MS: 799.2 [M + Na]+; Anal. calcd for C28H48N4O21: C, 43.30; H, 6.23; found: C, 43.14; H, 6.45.
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra of compounds 1 and 6–10. | ||
| Format: PDF | Size: 4.3 MB | Download |
References
-
Hisham, N. J. Trop. Pediatr. 1982, 28, 1–4.
Return to citation in text: [1] -
Vila, J.; Gene, A.; Vargas, M.; Gascon, J.; Latorre, C.; Jimenez De Anta, M. T. J. Med. Microbiol. 1998, 47, 889–891. doi:10.1099/00222615-47-10-889
Return to citation in text: [1] -
Kaper, J. B.; Nataro, J. P.; Mobley, H. L. T. Nat. Rev. Microbiol. 2004, 2, 123–140. doi:10.1038/nrmicro818
Return to citation in text: [1] -
Russo, T. A.; Johnson, J. R. J. Infect. Dis. 2000, 181, 1753–1754. doi:10.1086/315418
Return to citation in text: [1] -
Johnson, J. R. Clin. Microbiol. Rev. 1991, 4, 80–128.
Return to citation in text: [1] -
Stenutz, R.; Weintraub, A.; Widmalm, G. FEMS Microbiol. Rev. 2006, 30, 382–403. doi:10.1111/j.1574-6976.2006.00016.x
Return to citation in text: [1] -
Beutin, L.; Aleksic, S.; Zimmermann, S.; Gleier, K. Med. Microbiol. Immunol. 1994, 183, 13–21. doi:10.1007/BF00193627
Return to citation in text: [1] -
Zhao, G.; Perepelov, A. V.; Senchenkova, S. N.; Shashkov, A. S.; Feng, L.; Li, X.; Knirel, Y. A.; Wang, L. Carbohydr. Res. 2007, 342, 1275–1279. doi:10.1016/j.carres.2007.03.005
Return to citation in text: [1] -
Roy, R. Drug Discovery Today: Technol. 2004, 1, 327–336. doi:10.1016/j.ddtec.2004.10.005
Return to citation in text: [1] -
Pozsgay, V. Curr. Top. Med. Chem. 2008, 8, 126–140. doi:10.2174/156802608783378864
Return to citation in text: [1] -
Ada, G.; Isaacs, D. Clin. Microbiol. Infect. 2003, 9, 79–85. doi:10.1046/j.1469-0691.2003.00530.x
Return to citation in text: [1] -
Vliegenthart, J. F. FEBS Lett. 2006, 580, 2945–2950. doi:10.1016/j.febslet.2006.03.053
Return to citation in text: [1] -
Mukherjee, C.; Ranta, K.; Savolainen, J.; Leino, R. Eur. J. Org. Chem. 2012, 2957–2968. doi:10.1002/ejoc.201200041
Return to citation in text: [1] [2] [3] -
Mandal, P. K.; Misra, A. K. Bioorg. Chem. 2010, 38, 56–61. doi:10.1016/j.bioorg.2010.01.001
Return to citation in text: [1] -
Basu, S.; Pal, J. N. Carbohydr. Res. 1990, 208, 241–245. doi:10.1016/0008-6215(90)80103-A
Return to citation in text: [1] -
Kihlberg, J. O.; Leigh, D. A.; Bundle, D. R. J. Org. Chem. 1990, 55, 2860–2863. doi:10.1021/jo00296a055
Return to citation in text: [1] -
Mukhopadhyaya, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119
Return to citation in text: [1] [2] [3] [4] -
Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f
Return to citation in text: [1] [2] -
Fraser-Reid, B.; Lopez, J. C. Top. Curr. Chem. 2011, 301, 1–29. doi:10.1007/128_2010_105
Return to citation in text: [1] [2] -
Mandal, P. K.; McMurray, J. S. J. Org. Chem. 2007, 72, 6599–6601. doi:10.1021/jo0706123
Return to citation in text: [1] [2] -
Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007
Return to citation in text: [1] -
Panchadhayee, R.; Misra, A. K. Synlett 2010, 1193–1196. doi:10.1055/s-0029-1219798
Return to citation in text: [1] -
Bergonzi, M. C.; Catelani, G.; D'Andrea, F.; De Rensis, F. Carbohydr. Res. 1998, 311, 231–234. doi:10.1016/S0008-6215(98)00211-0
Return to citation in text: [1] -
Kanie, O.; Ito, Y.; Ogawa, T. J. Am. Chem. Soc. 1994, 116, 12073–12074. doi:10.1021/ja00105a066
Return to citation in text: [1] -
Lee, H.-H.; Schwartz, D. A.; Harris, J. F.; Carver, J. P.; Krepinsky, J. J. Can. J. Chem. 1986, 64, 1912–1918. doi:10.1139/v86-315
Return to citation in text: [1]
| 6. | Stenutz, R.; Weintraub, A.; Widmalm, G. FEMS Microbiol. Rev. 2006, 30, 382–403. doi:10.1111/j.1574-6976.2006.00016.x |
| 20. | Mandal, P. K.; McMurray, J. S. J. Org. Chem. 2007, 72, 6599–6601. doi:10.1021/jo0706123 |
| 4. | Russo, T. A.; Johnson, J. R. J. Infect. Dis. 2000, 181, 1753–1754. doi:10.1086/315418 |
| 5. | Johnson, J. R. Clin. Microbiol. Rev. 1991, 4, 80–128. |
| 13. | Mukherjee, C.; Ranta, K.; Savolainen, J.; Leino, R. Eur. J. Org. Chem. 2012, 2957–2968. doi:10.1002/ejoc.201200041 |
| 3. | Kaper, J. B.; Nataro, J. P.; Mobley, H. L. T. Nat. Rev. Microbiol. 2004, 2, 123–140. doi:10.1038/nrmicro818 |
| 17. | Mukhopadhyaya, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 18. | Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f |
| 2. | Vila, J.; Gene, A.; Vargas, M.; Gascon, J.; Latorre, C.; Jimenez De Anta, M. T. J. Med. Microbiol. 1998, 47, 889–891. doi:10.1099/00222615-47-10-889 |
| 19. | Fraser-Reid, B.; Lopez, J. C. Top. Curr. Chem. 2011, 301, 1–29. doi:10.1007/128_2010_105 |
| 13. | Mukherjee, C.; Ranta, K.; Savolainen, J.; Leino, R. Eur. J. Org. Chem. 2012, 2957–2968. doi:10.1002/ejoc.201200041 |
| 15. | Basu, S.; Pal, J. N. Carbohydr. Res. 1990, 208, 241–245. doi:10.1016/0008-6215(90)80103-A |
| 9. | Roy, R. Drug Discovery Today: Technol. 2004, 1, 327–336. doi:10.1016/j.ddtec.2004.10.005 |
| 10. | Pozsgay, V. Curr. Top. Med. Chem. 2008, 8, 126–140. doi:10.2174/156802608783378864 |
| 11. | Ada, G.; Isaacs, D. Clin. Microbiol. Infect. 2003, 9, 79–85. doi:10.1046/j.1469-0691.2003.00530.x |
| 12. | Vliegenthart, J. F. FEBS Lett. 2006, 580, 2945–2950. doi:10.1016/j.febslet.2006.03.053 |
| 16. | Kihlberg, J. O.; Leigh, D. A.; Bundle, D. R. J. Org. Chem. 1990, 55, 2860–2863. doi:10.1021/jo00296a055 |
| 8. | Zhao, G.; Perepelov, A. V.; Senchenkova, S. N.; Shashkov, A. S.; Feng, L.; Li, X.; Knirel, Y. A.; Wang, L. Carbohydr. Res. 2007, 342, 1275–1279. doi:10.1016/j.carres.2007.03.005 |
| 7. | Beutin, L.; Aleksic, S.; Zimmermann, S.; Gleier, K. Med. Microbiol. Immunol. 1994, 183, 13–21. doi:10.1007/BF00193627 |
| 14. | Mandal, P. K.; Misra, A. K. Bioorg. Chem. 2010, 38, 56–61. doi:10.1016/j.bioorg.2010.01.001 |
| 22. | Panchadhayee, R.; Misra, A. K. Synlett 2010, 1193–1196. doi:10.1055/s-0029-1219798 |
| 13. | Mukherjee, C.; Ranta, K.; Savolainen, J.; Leino, R. Eur. J. Org. Chem. 2012, 2957–2968. doi:10.1002/ejoc.201200041 |
| 21. | Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007 |
| 20. | Mandal, P. K.; McMurray, J. S. J. Org. Chem. 2007, 72, 6599–6601. doi:10.1021/jo0706123 |
| 18. | Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f |
| 17. | Mukhopadhyaya, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 25. | Lee, H.-H.; Schwartz, D. A.; Harris, J. F.; Carver, J. P.; Krepinsky, J. J. Can. J. Chem. 1986, 64, 1912–1918. doi:10.1139/v86-315 |
| 17. | Mukhopadhyaya, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 19. | Fraser-Reid, B.; Lopez, J. C. Top. Curr. Chem. 2011, 301, 1–29. doi:10.1007/128_2010_105 |
| 24. | Kanie, O.; Ito, Y.; Ogawa, T. J. Am. Chem. Soc. 1994, 116, 12073–12074. doi:10.1021/ja00105a066 |
| 17. | Mukhopadhyaya, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 23. | Bergonzi, M. C.; Catelani, G.; D'Andrea, F.; De Rensis, F. Carbohydr. Res. 1998, 311, 231–234. doi:10.1016/S0008-6215(98)00211-0 |
© 2012 Sau and Misra; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)