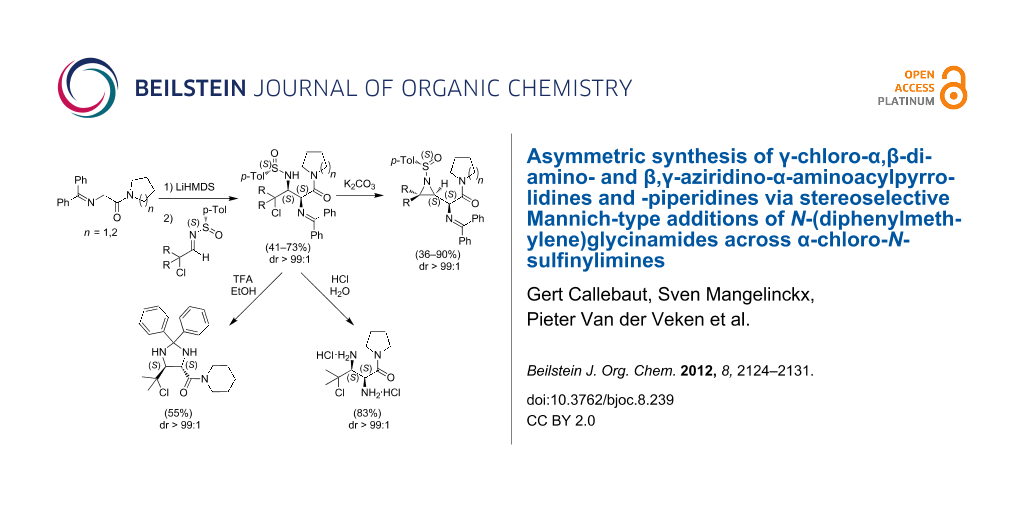
Abstract
The asymmetric synthesis of new chiral γ-chloro-α,β-diaminocarboxylamide derivatives by highly diastereoselective Mannich-type reactions of N-(diphenylmethylene)glycinamides across chiral α-chloro-N-p-toluenesulfinylaldimines was developed. The resulting (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides were formed with the opposite enantiotopic face selectivity as compared to the (SS,2R,3R)-γ-chloro-α,β-diaminocarboxyl esters obtained via Mannich-type addition of analogous N-(diphenylmethylene)glycine esters across a chiral α-chloro-N-p-toluenesulfinylaldimine. Selective deprotection under different acidic reaction conditions and ring closure of the γ-chloro-α,β-diaminocarboxylamides was optimized, which resulted in Nα-deprotected syn-γ-chloro-α,β-diaminocarboxylamides, N-sulfinyl-β,γ-aziridino-α-aminocarboxylamide derivatives, a trans-imidazolidine, and an Nα,Nβ-deprotected syn-γ-chloro-α,β-diaminocarboxylamide.

Graphical Abstract
Introduction
In recent years, non-proteinogenic diaminocarboxylic acids have gained a lot of attention among organic chemists and biochemists [1-3]. This is due to the fact that these diaminocarboxylic acids are present as key structural fragments in biologically active compounds, and some are also bioactive as the free diaminocarboxylic acid derivative [1-6]. For example, α,γ-diaminoacylamides are known for their high potency and selectivity as dipeptidyl peptidase (DPP) inhibitors [7-9].
DPP IVs are proteases that specifically cleave off N-terminal dipeptides and are involved in the degradation of incretin hormones, including glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). GLP-1 is involved in the regulation of glucose homeostasis through stimulation of insulin secretion, inhibition of glucagon release, and delay of gastric emptying. It has been demonstrated that the presence of intravenous GLP-1 increases insulin secretion as a response to elevated glucose levels, and as such, GLP-1 can offer therapeutic benefits for patients with type 2 diabetes. Unfortunately, therapeutic application of GLP-1 is problematic due to the lack of oral activity and the rapid degradation by plasma DPP IV. Therefore, DPP IV inhibitors could offer a solution to this problem, as they can extend the duration of action of GLP-1 and prolong the beneficial effects [10-12].
Besides DPP IV, a few related enzymes are present in the family of DPPs, with DPP II, DPP8, DPP9 and FAP being the most important regarding the therapeutic potential, when focusing on the inhibitory potency and selectivity [10-12]. In the research focused on DPP II and DPP IV inhibitors, it has been found that the α,γ-diaminoacylpiperidine, (S)-2,4-diaminobutanoylpiperidine, is a lead compound in the development of a large series of highly potent and selective DPP II inhibitors [7-9] (Figure 1). Next to the α,γ-diaminoacylpyrrolidines and –piperidines, which exhibit a DPP inhibitory effect, some β-aminocarboxylamides, such as sitagliptin, are also known as DPP inhibitors [13]. Sitagliptin is a commercialized oral antihyperglycemic drug of the DPP IV inhibitor class [14].
![[1860-5397-8-239-1]](/bjoc/content/figures/1860-5397-8-239-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
As α,γ-diaminocarboxylamides, as well as β-aminocarboxylamides, are known for their activity as DPP inhibitors, an increasing interest to study the DPP inhibitory potency of analogous α,β-diaminocarboxylamides exists [15]. The synthesis of chiral α,β-diaminocarboxylic acid derivatives by asymmetric Mannich-type addition of enolates across activated imines, e.g., N-sulfinylimines [16-20], is one of the most common and versatile methods in organic chemistry and is continuously under development [1-3]. Recently, our research group elaborated the asymmetric synthesis of new chiral γ-chloro-α,β-diaminocarboxyl esters by highly diastereoselective Mannich-type reactions of N-(diphenylmethylene)glycine esters across a chiral α-chloro-N-p-toluenesulfinylimine [20], which belongs to the useful class of α-halo-imines [21-26]. However, transformation of γ-chloro-α,β-diaminocarboxyl esters into the corresponding carboxylic acids, en route to further coupling to carboxylamides, has proven to be unsuccessful, probably due to competitive reactions such as the formation of α,β-diamino-γ-butyrolactones [20].
The results discussed within the present paper demonstrate the synthesis and elaboration of chiral syn-γ-chloro-α,β-diaminocarboxylamide derivatives with excellent diastereoselectivity. In order to develop potential DPP inhibitors, the ring closure and deprotection of the α-amino functionality of the synthesized γ-chloro-α,β-diaminocarboxylamides were explored as well.
Results and Discussion
The stereoselective synthesis of chiral γ-chloro-α,β-diaminocarboxylamides was performed by using a Mannich-type addition of glycine amides 4 across chiral α-chloro-N-sulfinylaldimines 3.
Initially, the chiral α-chloro-N-sulfinylaldimines 3, including the new imines 3b and 3c derived from 2-chloro-2-ethylbutanal (1b) and 1-chlorocyclohexanecarboxaldehyde (1c), respectively, were efficiently prepared by condensation of α-chloroaldehydes 1 with (S)-(+)-p-toluenesulfinamide (2) in dichloromethane in the presence of Ti(OEt)4 (Scheme 1) [27].
![[1860-5397-8-239-i1]](/bjoc/content/inline/1860-5397-8-239-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of chiral α-chloro-N-p-toluenesulfinylaldimines 3.
Scheme 1: Synthesis of chiral α-chloro-N-p-toluenesulfinylaldimines 3.
The synthesis of N-(diphenylmethylene)glycinamides 4 was performed starting from N-Boc glycine, in accordance with literature procedures [28,29]. Based on our previously reported Mannich-type addition of glycine esters across chiral α-chloro-N-p-toluenesulfinylaldimine 3a [20], the influence of the base (LiHMDS or LDA) used for the deprotonation of glycine amides 4 on the syn- or anti-selectivity of the Mannich-type addition was investigated (Scheme 2).
![[1860-5397-8-239-i2]](/bjoc/content/inline/1860-5397-8-239-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides 5. aYield in parentheses results from the use of LDA instead of LiHMDS.
Scheme 2: Synthesis of (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides 5. aYield in parentheses results from th...
Initially, the Mannich-type addition of glycine amide 4b across chiral α-chloro-N-p-toluenesulfinylisobutyraldimine (3a) was performed at −78 °C using 1.1 equiv of LDA. Upon 1H NMR analysis of the crude reaction mixture, the resulting syn-γ-chloro-α,β-diaminocarboxylamide syn-5b was formed with an excellent stereoselectivity (dr > 99:1) but the conversion was rather low. After crystallization, the syn-adduct syn-5b was isolated in a low yield of 16%. Repeating the Mannich-type addition of glycinamides 4 across chiral α-chloro-N-p-toluenesulfinylaldimines 3 with 1.1 equiv of LiHMDS resulted also in the formation of syn-γ-chloro-α,β-diaminocarboxylamides syn-5 with an excellent stereoselectivity (dr > 99:1). Because of the complete conversion of the substrates under these better reaction conditions (−78 °C, 15 min), the syn-adducts syn-5 could be isolated in higher yields (41–73%) after recrystallization. The diastereomeric ratio of these syn-γ-chloro-α,β-diaminocarboxylamides syn-5 (dr > 99:1) was determined by a combination of 1H NMR, 13C NMR and HPLC analysis in which no signals from other diastereomers could be detected.
In contrast to the Mannich-type addition of glycine esters across chiral α-chloro-N-p-toluenesulfinylimine 3a [20], the diastereoselectivity of the Mannich-type addition of glycinamides 4 across chiral α-chloro-N-p-toluenesulfinylaldimines 3 was independent of the base used. The absolute stereochemistry of (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides syn-5 was unambiguously determined by means of an X-ray diffraction analysis of compound syn-5b (Figure 2), in combination with the analogous NMR chemical shifts (Hα: δ = 4.91–5.20 ppm, Hβ: δ = 3.74–4.05 ppm) and the characteristic vicinal coupling constants (3JHα-Hβ = 0–1.1 Hz) of all derivatives syn-5a–f.
![[1860-5397-8-239-2]](/bjoc/content/figures/1860-5397-8-239-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of syn-γ-chloro-α,β-diaminocarboxylamide syn-5b.
Figure 2: Crystal structure of syn-γ-chloro-α,β-diaminocarboxylamide syn-5b.
The vicinal coupling constant 3JHα-Hβ = 0–1.1 Hz for the syn-amides 5 has a comparably small value as the observed vicinal coupling constant 3JHα-Hβ of the closely related syn-γ-chloro-α,β-diaminocarboxyl esters (3JHα-Hβ = 1.1 Hz) [20]. Notably, the (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides syn-5 were obtained with the opposite enantioselectivity as compared to the (SS,2R,3R)-γ-chloro-α,β-diaminocarboxyl esters obtained by Mannich-type addition of E-enolates derived from glycine esters across imines 3 [20].
The monosubstituted tertiary amide enolates obtained by deprotonation of N-(diphenylmethylene)glycinamides 4 are expected to have the Z-geometry in which A(1,3) interactions are minimized and Li-chelation stabilizes the conformation (Scheme 3), regardless of the base that was used [30,31].
![[1860-5397-8-239-i3]](/bjoc/content/inline/1860-5397-8-239-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Transition-state model for reaction of the Z-enolate of glycinamides 4 in the Mannich-type addition across chiral α-chloro-N-p-toluenesulfinyl aldimines 3.
Scheme 3: Transition-state model for reaction of the Z-enolate of glycinamides 4 in the Mannich-type addition...
Reaction of the Z-enolates via a cyclic chelated six-membered chairlike transition-state model TS-6a, would have resulted in anti-addition products anti-5 in analogy with our previously obtained results on the synthesis of (SS,2S,3R)-γ-chloro-α,β-diaminocarboxyl esters [20]. However, starting from glycinamides 4, due to the important 1,3-diaxial interaction between the haloalkyl group (–CClR2) and the cyclic amine moiety [–N(CH2)n] in this transition state, TS-6a is highly disfavored. The formation of the (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides syn-5 can be explained by a boatlike transition-state model TS-6b involving the (E)-N-p-toluenesulfinylaldimines 3 [32-35]. This less sterically hindered transition state TS-6b, in which the haloalkyl group (–CClR2) occupies the less hindered pseudoequatorial position, and the corresponding Li-adduct 7 are stabilized by the interaction between the Li-cation, the diphenylmethyleneamino group, and the sulfinylimine nitrogen.
The reversal of the enantiotopic face selectivity in the reaction of the N-sulfinylimines 3 with the glycinamides 4, as compared to the reaction with glycine esters, is attributed to the α-coordinating ability of the chlorine atom with the lithium of the incoming enolate as depicted in transition state TS-6b. The coordinating α-chloro atom in TS-6b overrides the chelation of the sulfinyl oxygen (e.g., TS-6a) and allows the sulfinylimine to react in the conformation wherein the S=O bond and the lone pair of electrons on the nitrogen atom are antiperiplanar [36]. This reversal of stereoselectivity is analogous to results obtained with other N-p-toluenesulfinylimines containing an oxygen atom as α-coordinating group [37-39]. The resulting syn-addition products syn-5 were subsequently cyclized to the corresponding N-sulfinyl-β,γ-aziridino-α-aminocarboxylamides 8 upon treatment with K2CO3 in acetone under reflux in a moderate to very good yield (36–90%, Scheme 4).
![[1860-5397-8-239-i4]](/bjoc/content/inline/1860-5397-8-239-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N-sulfinyl-β,γ-aziridino-α-amino carboxylic amides 8.
Scheme 4: Synthesis of N-sulfinyl-β,γ-aziridino-α-amino carboxylic amides 8.
The conversion of the ring-closure reaction was always complete as determined by TLC analysis, but purification of these N-sulfinyl-β,γ-aziridino-α-aminocarboxylamides 8 by flash chromatography resulted in a considerable loss of product.
In order to extend the potential applicability of the synthesized N-sulfinyl-β,γ-aziridino-α-aminocarboxylamides 8 as building blocks in biomedicinal chemistry, some attempts were made to remove the N-protective groups of diaminocarboxylamides 8 under mild acidic conditions (Scheme 4). In analogy with our recently published results on the corresponding aziridino esters [20], amide 8b was treated with 5 equiv of trifluoroacetic acid in acetone/water (2:1) at rt for 15 min. After a basic workup with NH4OH, it was concluded that the conversion towards the N-deprotected syn-β,γ-aziridino-α-aminocarboxylamide 9b was complete based on 1H NMR and LC–MS analysis of the crude reaction mixture. Unfortunately, all attempted purification techniques (column chromatography, preparative TLC, acid-base extraction) to remove benzophenone and some other minor impurities from the crude reaction mixture, failed to provide the pure N-deprotected syn-β,γ-aziridino-α-aminocarboxylamide 9b.
Alternatively, the deprotection of the α-amino functionality of the synthesized syn-γ-chloro-α,β-diaminocarboxylamides syn-5 was investigated en route towards the development of potential DPP inhibitors [7-9]. The syn-γ-chloro-α,β-diaminocarboxylamides syn-5 were treated with 5 equiv of trifluoroacetic acid in acetone/water (2:1) for 15 min (Scheme 5).
![[1860-5397-8-239-i5]](/bjoc/content/inline/1860-5397-8-239-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: α-Deprotection and subsequent ring-closure of syn-γ-chloro-α,β-diamino carboxylic amides syn-5.
Scheme 5: α-Deprotection and subsequent ring-closure of syn-γ-chloro-α,β-diamino carboxylic amides syn-5.
After a basic workup with NH4OH, the α-deprotected syn-γ-chloro-α,β-diaminocarboxylamides 10 could be purified by crystallization or preparative TLC (21–91% yield). The obtained result was in accordance with the earlier reported selective deprotection of a benzophenone imine functionality, in the presence of an N-p-toluenesulfinyl moiety, of diamino esters with H3PO4/H2O/THF [17,40].
In a subsequent step, syn-γ-chloro-α,β-diaminocarboxylamide 10b was chemoselectively cyclized to the corresponding N-sulfinyl-β,γ-aziridino-α-aminocarboxylamide 11b upon treatment with K2CO3 in acetone under reflux in 86% yield. In order to provide access to the Nα,Nβ-deprotected syn-γ-chloro-α,β-diaminocarboxylamides, syn-γ-chloro-α,β-diaminocarboxylamides syn-5 were subjected to some alternative acidic deprotection reactions (Scheme 6).
![[1860-5397-8-239-i6]](/bjoc/content/inline/1860-5397-8-239-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: N-p-toluenesulfinyl-deprotection of syn-γ-chloro-α,β-diaminocarboxylamides syn-5.
Scheme 6: N-p-toluenesulfinyl-deprotection of syn-γ-chloro-α,β-diaminocarboxylamides syn-5.
In an initial reaction, syn-γ-chloro-α,β-diaminocarboxylamide syn-5b was treated with 10 equiv of trifluoroacetic acid in ethanol at rt [18]. This resulted in trans-imidazolidine 12b after basic workup with NH4OH. It is remarkable that the N-(diphenylmethylene) group was not removed under these reaction conditions but was trapped by the deprotected β-amino group, as the deprotection of analogous anti-substrates under the same reaction conditions led to unprotected anti-α,β-diaminocarboxyl esters [18]. This is possibly due to the fact that solvolysis of the imine functionality with ethanol is not favorable and an acid-catalyzed deprotection of the sulfinyl moiety will occur first [41,42]. The resulting β-amino deprotected syn-γ-chloro-α,β-diaminocarboxylamide could subsequently ring close further to trans-imidazolidine 12b, which will be less sterically congested than an analogous cis-imidazolidine. In the literature, comparable non-halogenated trans-imidazolidines were already synthesized by 1,3-dipolar cycloaddition of N-benzylidene glycine ester enolates across N-sulfinylaldimines in the presence of a Lewis acid [43]. The trans-stereochemistry of imidazolidine 12b was ensured by the vicinal coupling constant 3JH4-H5 = 7.43 Hz and the 1H NMR chemical shift of H4 (3.85 ppm), which were in the same range as for closely related trans-imidazolidines and trans-oxazolidines [43-45]. The trans-imidazolidine 12b is a potential building block for foldamers, as the corresponding trans-oxazolidin-2-ones are already applied as such [46]. trans-Imidazolidine 12b could also be used as a precursor of the corresponding Nα,Nβ-deprotected α,β-diaminocarboxylamide, by hydrolysis under acidic conditions, in analogy with deprotection reactions of imidazolidines, imidazolines and oxazolines in the literature [16,47,48]. However, in a second reaction, syn-γ-chloro-α,β-diaminocarboxylamide syn-5a was directly converted into the dihydrochloride of the Nα,Nβ-deprotected syn-γ-chloro-α,β-diaminocarboxylamide 13a, by stirring in 0.5 M (aq) HCl/EtOAc (2:1) for 30 min at rt, in a yield of 83%. In this reaction, the acid-catalyzed hydrolysis of the benzophenone imine functionality proceeds readily and prevents the formation of the corresponding trans-imidazolidine.
Conclusion
In conclusion, it was demonstrated that new chiral syn-γ-chloro-α,β-diaminocarboxylamides are formed in acceptable to good yields and with excellent diastereomeric ratios by stereoselective Mannich-type reactions of N-(diphenylmethylene)glycinamides across chiral α-chloro-N-p-toluenesulfinylaldimines. Notably, a very high syn-diastereoselectivity was obtained in the synthesis of the (SS,2S,3S)-γ-chloro-α,β-diaminocarboxylamides with the opposite enantiotopic face selectivity as compared to the Mannich-type additions of N-(diphenylmethylene)glycine esters across chiral α-chloro-N-p-toluenesulfinylaldimines. The synthesized γ-chloro-α,β-diaminocarboxylamides were selectively deprotected under acidic conditions, and the resulting α,β-diaminoacylpyrrolidines and -piperidines have a potential applicability as dipeptidyl peptidase inhibitors, which is currently under investigation.
Supporting Information
| Supporting Information File 1: General experimental conditions, experimental procedures and data, copies of 1H NMR and 13C NMR spectra for compounds 3, syn-5, 8, and 10–13. | ||
| Format: PDF | Size: 1.6 MB | Download |
| Supporting Information File 2: CIF-file of compound syn-5b. | ||
| Format: CIF | Size: 21.7 KB | Download |
References
-
Viso, A.; Fernández de la Pradilla, R.; Garcia, A.; Flores, A. Chem. Rev. 2005, 105, 3167. doi:10.1021/cr0406561
Return to citation in text: [1] [2] [3] -
Viso, A.; Fernández de la Pradilla, R.; Tortosa, M.; García, A.; Flores, A. Chem. Rev. 2011, 111, PR1. doi:10.1021/cr100127y
Return to citation in text: [1] [2] [3] -
Arrayás, R. G.; Carretero, J. C. Chem. Soc. Rev. 2009, 38, 1940. doi:10.1039/b820303b
Return to citation in text: [1] [2] [3] -
Blasiak, L. C.; Vaillancourt, F. H.; Walsh, C. T.; Drennan, C. L. Nature 2006, 440, 368. doi:10.1038/nature04544
Return to citation in text: [1] -
Vértesy, L.; Ehlers, E.; Kogler, H.; Kurz, M.; Meiwes, J.; Seibert, G.; Vogel, M.; Hammann, P. J. Antibiot. 2000, 53, 816. doi:10.7164/antibiotics.53.816
Return to citation in text: [1] -
Harrison, L.; Teplow, D. B.; Rinaldi, M.; Strobel, G. J. Gen. Microbiol. 1991, 137, 2857. doi:10.1099/00221287-137-12-2857
Return to citation in text: [1] -
Senten, K.; Van der Veken, P.; Bal, G.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Bauvois, B.; Haemers, A.; Augustyns, K. Bioorg. Med. Chem. Lett. 2002, 12, 2825. doi:10.1016/S0960-894X(02)00603-0
Return to citation in text: [1] [2] [3] -
Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2003, 46, 5005. doi:10.1021/jm0308803
Return to citation in text: [1] [2] [3] -
Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2004, 47, 2906. doi:10.1021/jm031122f
Return to citation in text: [1] [2] [3] -
Chen, S.-J.; Jiaang, W.-T. Curr. Top. Med. Chem. 2011, 11, 1447. doi:10.2174/156802611795860933
Return to citation in text: [1] [2] -
Van der Veken, P.; Haemers, A.; Augustyns, K. Curr. Top. Med. Chem. 2007, 7, 621. doi:10.2174/156802607780091046
Return to citation in text: [1] [2] -
Augustyns, K.; Van der Veken, P.; Senten, K.; Haemers, A. Curr. Med. Chem. 2005, 12, 971. doi:10.2174/0929867053507298
Return to citation in text: [1] [2] -
Scheen, A. J. Expert Opin. Pharmacother. 2012, 13, 81. doi:10.1517/14656566.2012.642866
Return to citation in text: [1] -
Baetta, R.; Corsini, A. Drugs 2011, 71, 1441. doi:10.2165/11591400-000000000-00000
Return to citation in text: [1] -
Scharpe, S.; Augustyns, K.; Haemers, A.; Lambeir, A.-M.; De Meester, I.; Senten, K.; Van der Veken, P. Preparation of aminoacyl piperidides and related compounds as dipeptidyl peptidase inhibitors. WO Pat. Appl. WO 2004076433 (A1), Sept 10, 2004.
Return to citation in text: [1] -
Viso, A.; Fernández de la Pradilla, R.; López-Rodríguez, M. L.; García, A.; Flores, A.; Alonso, M. J. Org. Chem. 2004, 69, 1542. doi:10.1021/jo035613j
Return to citation in text: [1] [2] -
Viso, A.; Fernández de la Pradilla, R.; Flores, A.; García, A.; Tortosa, M.; López-Rodríguez, M. L. J. Org. Chem. 2006, 71, 1442. doi:10.1021/jo052077h
Return to citation in text: [1] [2] -
Davis, F. A.; Deng, J. Org. Lett. 2004, 6, 2789. doi:10.1021/ol048981y
Return to citation in text: [1] [2] [3] -
Davis, F. A.; Zhang, Y.; Qiu, H. Org. Lett. 2007, 9, 833. doi:10.1021/ol063058c
Return to citation in text: [1] -
Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
De Kimpe, N.; Schamp, N. Org. Prep. Proced. Int. 1979, 11, 115. doi:10.1080/00304947909458134
Return to citation in text: [1] -
De Kimpe, N.; Sulmon, P.; Verhé, R.; De Buyck, L.; Schamp, N. J. Org. Chem. 1983, 48, 4320. doi:10.1021/jo00171a033
Return to citation in text: [1] -
De Kimpe, N.; Sulmon, P.; Brunet, P. J. Org. Chem. 1990, 55, 5777. doi:10.1021/jo00309a023
Return to citation in text: [1] -
Denolf, B.; Mangelinckx, S.; Törnroos, K.; De Kimpe, N. Org. Lett. 2006, 8, 3129. doi:10.1021/ol0611245
Return to citation in text: [1] -
Denolf, B.; Leemans, E.; De Kimpe, N. J. Org. Chem. 2007, 72, 3211. doi:10.1021/jo0624795
Return to citation in text: [1] -
Denolf, B.; Leemans, E.; De Kimpe, N. J. Org. Chem. 2008, 73, 5662. doi:10.1021/jo801077f
Return to citation in text: [1] -
Davis, F. A.; Zhang, Y.; Andemichael, Y.; Fang, T.; Fanelli, D. L.; Zhang, H. J. Org. Chem. 1999, 64, 1403. doi:10.1021/jo9820622
Return to citation in text: [1] -
Venuti, M. C.; Alvarez, R.; Bruno, J. J.; Strosberg, A. M.; Gu, L.; Chiang, H. S.; Massey, I. J.; Chu, N.; Fried, J. H. J. Med. Chem. 1988, 31, 2145. doi:10.1021/jm00119a015
Return to citation in text: [1] -
O’Donnell, M. J.; Polt, R. L. J. Org. Chem. 1982, 47, 2663. doi:10.1021/jo00134a030
Return to citation in text: [1] -
Manthorpe, J. M.; Gleason, J. L. J. Am. Chem. Soc. 2001, 123, 2091. doi:10.1021/ja0058280
Return to citation in text: [1] -
Evans, D. A.; Takacs, J. M. Tetrahedron Lett. 1980, 21, 4233. doi:10.1016/S0040-4039(00)92870-3
Return to citation in text: [1] -
Bernardi, A.; Gennari, C.; Raimondi, L.; Villa, M. B. Tetrahedron 1997, 53, 7705. doi:10.1016/S0040-4020(97)00435-3
Return to citation in text: [1] -
Silveira, C. C.; Vieira, A. S.; Braga, A. L.; Russowsky, D. Tetrahedron 2005, 61, 9312. doi:10.1016/j.tet.2005.07.058
Return to citation in text: [1] -
Kiss, L.; Mangelinckx, S.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. J. Org. Chem. 2007, 72, 7199. doi:10.1021/jo0710634
Return to citation in text: [1] -
Mangelinckx, S.; De Sterck, B.; Colpaert, F.; Catak, S.; Jacobs, J.; Rooryck, S.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N. J. Org. Chem. 2012, 77, 3415. doi:10.1021/jo300203t
Return to citation in text: [1] -
Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893. doi:10.1021/jo071139w
Return to citation in text: [1] -
Fujisawa, T.; Kooriyama, Y.; Shimizu, M. Tetrahedron Lett. 1996, 37, 3881. doi:10.1016/0040-4039(96)00707-1
Return to citation in text: [1] -
Koriyama, Y.; Nozawa, A.; Hayakawa, R.; Shimizu, M. Tetrahedron 2002, 58, 9621. doi:10.1016/S0040-4020(02)01250-4
Return to citation in text: [1] -
Morton, D.; Stockman, R. A. Tetrahedron 2006, 62, 8869. doi:10.1016/j.tet.2006.06.107
Return to citation in text: [1] -
Davis, F. A.; Zhang, Y. Tetrahedron Lett. 2009, 50, 5205. doi:10.1016/j.tetlet.2009.06.125
Return to citation in text: [1] -
Mikołajczyk, M.; Drabowicz, J.; Bujnicki, B. J. Chem. Soc., Chem. Commun. 1976, 568. doi:10.1039/C39760000568
Return to citation in text: [1] -
Mikołajczyk, M.; Drabowicz, J.; Bujnicki, B. Tetrahedron Lett. 1985, 26, 5699. doi:10.1016/S0040-4039(01)80924-2
Return to citation in text: [1] -
Viso, A.; Fernández de la Pradilla, R.; García, A.; Guerrero-Strachan, C.; Alonso, M.; Tortosa, M.; Flores, A.; Martínez-Ripoll, M.; Fonseca, I.; André, I.; Rodríguez, A. Chem.–Eur. J. 2003, 9, 2867. doi:10.1002/chem.200204674
Return to citation in text: [1] [2] -
Szöllősy, A.; Tischer, T.; Kádas, I.; Tőke, L.; Tóth, G. Tetrahedron 1999, 55, 7279. doi:10.1016/S0040-4020(99)00354-3
Return to citation in text: [1] -
Ooi, T.; Kameda, M.; Taniguchi, M.; Maruoka, K. J. Am. Chem. Soc. 2004, 126, 9685. doi:10.1021/ja048865q
Return to citation in text: [1] -
Tomasini, C.; Angelici, G.; Castellucci, N. Eur. J. Org. Chem. 2011, 3648. doi:10.1002/ejoc.201100493
Return to citation in text: [1] -
Hiyama, T.; Koide, H.; Fujita, S.; Nozaki, H. Tetrahedron 1973, 29, 3137. doi:10.1016/S0040-4020(01)93455-6
Return to citation in text: [1] -
Papa, C.; Tomasini, C. Eur. J. Org. Chem. 2000, 1569. doi:10.1002/(SICI)1099-0690(200004)2000:8<1569::AID-EJOC1569>3.0.CO;2-X
Return to citation in text: [1]
| 17. | Viso, A.; Fernández de la Pradilla, R.; Flores, A.; García, A.; Tortosa, M.; López-Rodríguez, M. L. J. Org. Chem. 2006, 71, 1442. doi:10.1021/jo052077h |
| 40. | Davis, F. A.; Zhang, Y. Tetrahedron Lett. 2009, 50, 5205. doi:10.1016/j.tetlet.2009.06.125 |
| 1. | Viso, A.; Fernández de la Pradilla, R.; Garcia, A.; Flores, A. Chem. Rev. 2005, 105, 3167. doi:10.1021/cr0406561 |
| 2. | Viso, A.; Fernández de la Pradilla, R.; Tortosa, M.; García, A.; Flores, A. Chem. Rev. 2011, 111, PR1. doi:10.1021/cr100127y |
| 3. | Arrayás, R. G.; Carretero, J. C. Chem. Soc. Rev. 2009, 38, 1940. doi:10.1039/b820303b |
| 10. | Chen, S.-J.; Jiaang, W.-T. Curr. Top. Med. Chem. 2011, 11, 1447. doi:10.2174/156802611795860933 |
| 11. | Van der Veken, P.; Haemers, A.; Augustyns, K. Curr. Top. Med. Chem. 2007, 7, 621. doi:10.2174/156802607780091046 |
| 12. | Augustyns, K.; Van der Veken, P.; Senten, K.; Haemers, A. Curr. Med. Chem. 2005, 12, 971. doi:10.2174/0929867053507298 |
| 27. | Davis, F. A.; Zhang, Y.; Andemichael, Y.; Fang, T.; Fanelli, D. L.; Zhang, H. J. Org. Chem. 1999, 64, 1403. doi:10.1021/jo9820622 |
| 10. | Chen, S.-J.; Jiaang, W.-T. Curr. Top. Med. Chem. 2011, 11, 1447. doi:10.2174/156802611795860933 |
| 11. | Van der Veken, P.; Haemers, A.; Augustyns, K. Curr. Top. Med. Chem. 2007, 7, 621. doi:10.2174/156802607780091046 |
| 12. | Augustyns, K.; Van der Veken, P.; Senten, K.; Haemers, A. Curr. Med. Chem. 2005, 12, 971. doi:10.2174/0929867053507298 |
| 28. | Venuti, M. C.; Alvarez, R.; Bruno, J. J.; Strosberg, A. M.; Gu, L.; Chiang, H. S.; Massey, I. J.; Chu, N.; Fried, J. H. J. Med. Chem. 1988, 31, 2145. doi:10.1021/jm00119a015 |
| 29. | O’Donnell, M. J.; Polt, R. L. J. Org. Chem. 1982, 47, 2663. doi:10.1021/jo00134a030 |
| 7. | Senten, K.; Van der Veken, P.; Bal, G.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Bauvois, B.; Haemers, A.; Augustyns, K. Bioorg. Med. Chem. Lett. 2002, 12, 2825. doi:10.1016/S0960-894X(02)00603-0 |
| 8. | Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2003, 46, 5005. doi:10.1021/jm0308803 |
| 9. | Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2004, 47, 2906. doi:10.1021/jm031122f |
| 21. | De Kimpe, N.; Schamp, N. Org. Prep. Proced. Int. 1979, 11, 115. doi:10.1080/00304947909458134 |
| 22. | De Kimpe, N.; Sulmon, P.; Verhé, R.; De Buyck, L.; Schamp, N. J. Org. Chem. 1983, 48, 4320. doi:10.1021/jo00171a033 |
| 23. | De Kimpe, N.; Sulmon, P.; Brunet, P. J. Org. Chem. 1990, 55, 5777. doi:10.1021/jo00309a023 |
| 24. | Denolf, B.; Mangelinckx, S.; Törnroos, K.; De Kimpe, N. Org. Lett. 2006, 8, 3129. doi:10.1021/ol0611245 |
| 25. | Denolf, B.; Leemans, E.; De Kimpe, N. J. Org. Chem. 2007, 72, 3211. doi:10.1021/jo0624795 |
| 26. | Denolf, B.; Leemans, E.; De Kimpe, N. J. Org. Chem. 2008, 73, 5662. doi:10.1021/jo801077f |
| 16. | Viso, A.; Fernández de la Pradilla, R.; López-Rodríguez, M. L.; García, A.; Flores, A.; Alonso, M. J. Org. Chem. 2004, 69, 1542. doi:10.1021/jo035613j |
| 47. | Hiyama, T.; Koide, H.; Fujita, S.; Nozaki, H. Tetrahedron 1973, 29, 3137. doi:10.1016/S0040-4020(01)93455-6 |
| 48. | Papa, C.; Tomasini, C. Eur. J. Org. Chem. 2000, 1569. doi:10.1002/(SICI)1099-0690(200004)2000:8<1569::AID-EJOC1569>3.0.CO;2-X |
| 1. | Viso, A.; Fernández de la Pradilla, R.; Garcia, A.; Flores, A. Chem. Rev. 2005, 105, 3167. doi:10.1021/cr0406561 |
| 2. | Viso, A.; Fernández de la Pradilla, R.; Tortosa, M.; García, A.; Flores, A. Chem. Rev. 2011, 111, PR1. doi:10.1021/cr100127y |
| 3. | Arrayás, R. G.; Carretero, J. C. Chem. Soc. Rev. 2009, 38, 1940. doi:10.1039/b820303b |
| 4. | Blasiak, L. C.; Vaillancourt, F. H.; Walsh, C. T.; Drennan, C. L. Nature 2006, 440, 368. doi:10.1038/nature04544 |
| 5. | Vértesy, L.; Ehlers, E.; Kogler, H.; Kurz, M.; Meiwes, J.; Seibert, G.; Vogel, M.; Hammann, P. J. Antibiot. 2000, 53, 816. doi:10.7164/antibiotics.53.816 |
| 6. | Harrison, L.; Teplow, D. B.; Rinaldi, M.; Strobel, G. J. Gen. Microbiol. 1991, 137, 2857. doi:10.1099/00221287-137-12-2857 |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 15. | Scharpe, S.; Augustyns, K.; Haemers, A.; Lambeir, A.-M.; De Meester, I.; Senten, K.; Van der Veken, P. Preparation of aminoacyl piperidides and related compounds as dipeptidyl peptidase inhibitors. WO Pat. Appl. WO 2004076433 (A1), Sept 10, 2004. |
| 1. | Viso, A.; Fernández de la Pradilla, R.; Garcia, A.; Flores, A. Chem. Rev. 2005, 105, 3167. doi:10.1021/cr0406561 |
| 2. | Viso, A.; Fernández de la Pradilla, R.; Tortosa, M.; García, A.; Flores, A. Chem. Rev. 2011, 111, PR1. doi:10.1021/cr100127y |
| 3. | Arrayás, R. G.; Carretero, J. C. Chem. Soc. Rev. 2009, 38, 1940. doi:10.1039/b820303b |
| 43. | Viso, A.; Fernández de la Pradilla, R.; García, A.; Guerrero-Strachan, C.; Alonso, M.; Tortosa, M.; Flores, A.; Martínez-Ripoll, M.; Fonseca, I.; André, I.; Rodríguez, A. Chem.–Eur. J. 2003, 9, 2867. doi:10.1002/chem.200204674 |
| 44. | Szöllősy, A.; Tischer, T.; Kádas, I.; Tőke, L.; Tóth, G. Tetrahedron 1999, 55, 7279. doi:10.1016/S0040-4020(99)00354-3 |
| 45. | Ooi, T.; Kameda, M.; Taniguchi, M.; Maruoka, K. J. Am. Chem. Soc. 2004, 126, 9685. doi:10.1021/ja048865q |
| 14. | Baetta, R.; Corsini, A. Drugs 2011, 71, 1441. doi:10.2165/11591400-000000000-00000 |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 46. | Tomasini, C.; Angelici, G.; Castellucci, N. Eur. J. Org. Chem. 2011, 3648. doi:10.1002/ejoc.201100493 |
| 13. | Scheen, A. J. Expert Opin. Pharmacother. 2012, 13, 81. doi:10.1517/14656566.2012.642866 |
| 41. | Mikołajczyk, M.; Drabowicz, J.; Bujnicki, B. J. Chem. Soc., Chem. Commun. 1976, 568. doi:10.1039/C39760000568 |
| 42. | Mikołajczyk, M.; Drabowicz, J.; Bujnicki, B. Tetrahedron Lett. 1985, 26, 5699. doi:10.1016/S0040-4039(01)80924-2 |
| 7. | Senten, K.; Van der Veken, P.; Bal, G.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Bauvois, B.; Haemers, A.; Augustyns, K. Bioorg. Med. Chem. Lett. 2002, 12, 2825. doi:10.1016/S0960-894X(02)00603-0 |
| 8. | Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2003, 46, 5005. doi:10.1021/jm0308803 |
| 9. | Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2004, 47, 2906. doi:10.1021/jm031122f |
| 16. | Viso, A.; Fernández de la Pradilla, R.; López-Rodríguez, M. L.; García, A.; Flores, A.; Alonso, M. J. Org. Chem. 2004, 69, 1542. doi:10.1021/jo035613j |
| 17. | Viso, A.; Fernández de la Pradilla, R.; Flores, A.; García, A.; Tortosa, M.; López-Rodríguez, M. L. J. Org. Chem. 2006, 71, 1442. doi:10.1021/jo052077h |
| 18. | Davis, F. A.; Deng, J. Org. Lett. 2004, 6, 2789. doi:10.1021/ol048981y |
| 19. | Davis, F. A.; Zhang, Y.; Qiu, H. Org. Lett. 2007, 9, 833. doi:10.1021/ol063058c |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 43. | Viso, A.; Fernández de la Pradilla, R.; García, A.; Guerrero-Strachan, C.; Alonso, M.; Tortosa, M.; Flores, A.; Martínez-Ripoll, M.; Fonseca, I.; André, I.; Rodríguez, A. Chem.–Eur. J. 2003, 9, 2867. doi:10.1002/chem.200204674 |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 7. | Senten, K.; Van der Veken, P.; Bal, G.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Bauvois, B.; Haemers, A.; Augustyns, K. Bioorg. Med. Chem. Lett. 2002, 12, 2825. doi:10.1016/S0960-894X(02)00603-0 |
| 8. | Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2003, 46, 5005. doi:10.1021/jm0308803 |
| 9. | Senten, K.; Van der Veken, P.; De Meester, I.; Lambeir, A.-M.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2004, 47, 2906. doi:10.1021/jm031122f |
| 36. | Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893. doi:10.1021/jo071139w |
| 37. | Fujisawa, T.; Kooriyama, Y.; Shimizu, M. Tetrahedron Lett. 1996, 37, 3881. doi:10.1016/0040-4039(96)00707-1 |
| 38. | Koriyama, Y.; Nozawa, A.; Hayakawa, R.; Shimizu, M. Tetrahedron 2002, 58, 9621. doi:10.1016/S0040-4020(02)01250-4 |
| 39. | Morton, D.; Stockman, R. A. Tetrahedron 2006, 62, 8869. doi:10.1016/j.tet.2006.06.107 |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 32. | Bernardi, A.; Gennari, C.; Raimondi, L.; Villa, M. B. Tetrahedron 1997, 53, 7705. doi:10.1016/S0040-4020(97)00435-3 |
| 33. | Silveira, C. C.; Vieira, A. S.; Braga, A. L.; Russowsky, D. Tetrahedron 2005, 61, 9312. doi:10.1016/j.tet.2005.07.058 |
| 34. | Kiss, L.; Mangelinckx, S.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. J. Org. Chem. 2007, 72, 7199. doi:10.1021/jo0710634 |
| 35. | Mangelinckx, S.; De Sterck, B.; Colpaert, F.; Catak, S.; Jacobs, J.; Rooryck, S.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N. J. Org. Chem. 2012, 77, 3415. doi:10.1021/jo300203t |
| 20. | Callebaut, G.; Mangelinckx, S.; Kiss, L.; Sillanpää, R.; Fülöp, F.; De Kimpe, N. Org. Biomol. Chem. 2012, 10, 2326. doi:10.1039/c2ob06637h |
| 30. | Manthorpe, J. M.; Gleason, J. L. J. Am. Chem. Soc. 2001, 123, 2091. doi:10.1021/ja0058280 |
| 31. | Evans, D. A.; Takacs, J. M. Tetrahedron Lett. 1980, 21, 4233. doi:10.1016/S0040-4039(00)92870-3 |
© 2012 Callebaut et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)