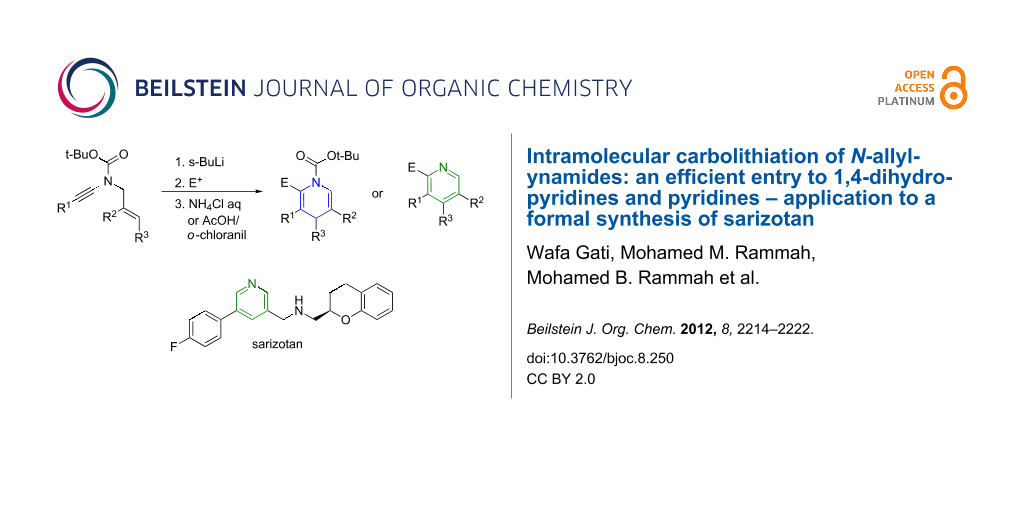
Abstract
We have developed a general synthesis of polysubstituted 1,4-dihydropyridines and pyridines based on a highly regioselective lithiation/6-endo-dig intramolecular carbolithiation from readily available N-allyl-ynamides. This reaction, which has been successfully applied to the formal synthesis of the anti-dyskinesia agent sarizotan, further extends the use of ynamides in organic synthesis and further demonstrates the synthetic efficiency of carbometallation reactions.

Graphical Abstract
Introduction
Since the discovery of the carbometallation reaction by Ziegler and Bähr in 1928 [1], this reaction has evolved as a most powerful tool to construct carbon–carbon bonds. An ever increasing number of organometallic species have been shown over the years to be suitable reagents for the carbometallation of various carbon–carbon multiple bonds. Lithium, copper, zinc, magnesium, zirconium, titanium, palladium and other metals are suitable for this transformation and considerable progress has recently been made in this area. Among these systems, the carbometallation of alkynes constitutes a most efficient entry to polysubstituted alkenes, provided that both the regioselectivity and the stereoselectivity can be controlled [2-5]. In this context, an efficient strategy to control these selectivity issues is the incorporation of a heteroatom directly attached to the triple bond, which can dramatically affect both the regio- and stereochemical outcomes due to the polarization of the triple bond and/or the formation of chelation-stabilized vinylmetal species. The carbometallation of O-, N-, P-, S-, and Si-substituted alkynes has indeed been quite extensively studied and shown to provide most efficient entries to polysubstituted, stereodefined heteroatom-substituted alkenes and has been implemented in remarkably elegant processes [6]. Intramolecular versions are especially attractive and provide most useful entries to highly substituted carbo- and heterocycles.
Based on our recent interest in the chemistry of ynamides [7-18] and inspired by recent reports from Meyer and Cossy [19-21], Marek [22-24] and Lam [25,26] on their carbopalladation, carbocupration and carbozincation, respectively, we decided to study the intramolecular carbolithiation of ynamides, which may provide an interesting entry to highly functionalized 1,4-dihydropyridines [27-29] and pyridines [30-33], most useful building blocks in organic synthesis and medicinal chemistry as well. Our strategy is summarized in Scheme 1 and is based on the following assumptions: According to the remarkable work of the Beak group on the α-lithiation of Boc-protected amines [34-38], N-allyl-ynamides 1 should be readily deprotonated to afford a transient chelation-stabilized allyllithium 2 and, provided that a metallotropic equilibrium exists between this intermediate and the less-stable allyllithium 3, an intramolecular carbometallation may then occur to yield a chelation-stabilized vinyllithium 4 and drive the overall process to the formation of the heterocyclic ring system. Further reaction with an electrophile followed by aqueous workup or hydrolysis under acidic and oxidative conditions would then afford the highly substituted dihydropyridine or pyridine derivatives 5 and 6, respectively. While there were no examples of such exclusive anionic 6-endo-dig cyclizations reported to the best of our knowledge [39], we felt that the presence of the chelating group on the nitrogen may allow such a selective process and a clean formation of the (dihydro)pyridine ring system. We have indeed demonstrated the efficiency of this strategy [40] and now report in this manuscript a full account on this work as well as the application of our pyridine synthesis to a formal synthesis of the anti-dyskinesia agent sarizotan.
![[1860-5397-8-250-i1]](/bjoc/content/inline/1860-5397-8-250-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Strategy for the synthesis of (1,4-dihydro)pyridines by deprotonation/intramolecular carbolithiation.
Scheme 1: Strategy for the synthesis of (1,4-dihydro)pyridines by deprotonation/intramolecular carbolithiatio...
Results and Discussion
Feasibility of the deprotonation/intramolecular carbolithiation
To first evaluate the compatibility of the ynamide moiety with the lithiation step and address potential problems associated with competitive carbolithiation of the activated alkyne, we first reacted N-benzyl-ynamide 7 with one equivalent of sec-butyllithium and tetramethylethylenediamine (TMEDA) in THF at −78 °C for 15 minutes, followed by the addition of methyl iodide. The corresponding N-phenylethyl-ynamide 8 was obtained in nearly quantitative yield, therefore demonstrating the compatibility of the ynamide group with the deprotonation step (Scheme 2a), although longer reaction times before the addition of methyl iodide resulted in much lower yields and extensive degradation: The intramolecular carbolithiation of N-allyl-ynamides to 1,4-dihydropyridines might therefore be possible, provided that the overall reaction rate is not too slow. To further test this hypothesis, N-allyl-ynamide 1a was reacted under similar reaction conditions and to our delight, a smooth cyclization occurred, the expected 1,4-dihydropyridine 5a being virtually formed as virtually the sole product (Scheme 2b), a clean reaction being a strict requirement for the development of a general route to 1,4-dihydropyridines devoided of an electron-withdrawing group at C-3, due to the instability of these acid- and oxygen-sensitive molecules. In situ conversion of the intermediate dihydropyridine to the corresponding pyridine by replacing the saturated aqueous ammonium chloride solution, used for the workup, by a combination of acetic acid and o-chloranil [41] was equally successful and provided the expected pyridine 6a in 81% yield (Scheme 2c).
![[1860-5397-8-250-i2]](/bjoc/content/inline/1860-5397-8-250-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Feasibility of the deprotonation/intramolecular carbolithiation.
Scheme 2: Feasibility of the deprotonation/intramolecular carbolithiation.
Synthesis of the starting N-allyl-ynamides
Before moving to the evaluation of the scope and limitations of this intramolecular carbolithiation, we had to prepare a set of ynamides possessing representative substituents on both the ynamide and allyl groups. Among all the methods evaluated, Hsung’s second-generation synthesis based on the copper-mediated cross-coupling between bromoalkynes 9 and nitrogen nucleophiles [42] turned out to be the most convenient one, the use of terminal alkynes [43], gem-dibromoalkenes [7], potassium alkynyltrifluoroborates [8] or copper acetylides [12] being less efficient when bulky N-Boc-allylamines 10 were used as nucleophiles. By using a slightly modified Hsung’s procedure, a series of N-allyl-ynamides 1 could be readily prepared in acceptable yields using a combination of copper(II) sulfate pentahydrate (40 mol %) and 1,10-phenanthroline (80 mol %) with potassium phosphate in refluxing toluene, the major side reaction observed in all cases being the competitive dimerization of the starting bromoalkynes (Scheme 3).
![[1860-5397-8-250-i3]](/bjoc/content/inline/1860-5397-8-250-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the starting N-allyl-ynamides.
Scheme 3: Synthesis of the starting N-allyl-ynamides.
Intramolecular carbolithiation of N-allyl-ynamides to 1,4-dihydropyridines and pyridines: scope and limitations
With this set of ynamides in hand, we next evaluated their cyclization to the corresponding 1,4-dihydropyridines 5 or pyridines 6 using the lithiation/intramolecular carbolithiation sequence. All N-allyl-ynamides 1 shown in Scheme 3 were therefore treated with s-butyllithium and TMEDA in THF at −78 °C for one hour followed by the addition of an aqueous saturated solution of ammonium chloride (Scheme 4, conditions A) or acetic acid and o-chloranil (Scheme 4, conditions B), yielding the corresponding 1,4-dihydropyridines 5 or pyridines 6, respectively. Results from these studies are collected in Scheme 4, yields being indicated only in the pyridine series due to the high sensitivity of the 1,4-dihydropyridine derivatives, which were obtained in virtually pure form in crude reaction mixtures. As evidenced with these results, 3-aryl-1,4-dihydropyridines and pyridines (5/6a–f and l–r) are readily obtained from the corresponding aryl-substituted N-allyl-ynamides regardless of the substitution pattern or the electronic properties of the aromatic ring. The reaction is also efficient in the case of an alkenyl group (5/6g) and the presence of an alkyl group was, as expected, more problematic due to competitive propargylic lithiation. Indeed, while a tert-butyl group was well tolerated (5/6h), the presence of secondary or primary alkyl chains such as cyclohexyl (5/6i) or n-hexyl (5/6j) groups did not afford the cyclized products, which could also not be obtained either when starting from a TIPS-protected primary ynamide (5/6k), the silicon protecting group being readily cleaved under the reaction conditions. The influence of the substitution pattern of the allyl moiety was next carefully examined and substitution at the β-, γ-positions, or both, was well tolerated, yielding the 3,5-disubstituted- (5/6l–o), 3,4-disubstituted- (5/6p–q) and 3,4,5-trisubstituted- (5/6r) 1,4-dihydropyridines and pyridines in good yields, respectively, compounds that are rather challenging to obtain using other synthetic routes. In addition, the reaction can be performed on a gram-scale with similar efficiency. This was indeed briefly evaluated for the anionic cyclization of ynamide 1a and the exact same yield of the corresponding pyridine 6a (81%) was obtained on a gram scale.
![[1860-5397-8-250-i4]](/bjoc/content/inline/1860-5397-8-250-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Intramolecular carbolithiation of N-allyl-ynamides to 1,4-dihydropyridines and pyridines.
Scheme 4: Intramolecular carbolithiation of N-allyl-ynamides to 1,4-dihydropyridines and pyridines.
With a chelation-stabilized vinyllithium being formed after the anionic 6-endo-dig cyclization, we next considered the possibility of trapping this vinyllithium with an electrophile, which might allow the introduction of an additional C-2 substituent. N-Allyl-ynamide 1a was therefore cyclized under our standard conditions and then treated with deuterated water (Scheme 5a) or methyl iodide (Scheme 5b) before the acidic and oxidative workup. While the desired 2,3-disubstituted 6s and 6t were indeed formed under these conditions, they could only be isolated in modest yields (30–33%), even in the presence of additional HMPA, which might may constitute the major limitation of our process. Other attempts involving electrophiles such as acid chlorides and allyl bromide or transmetallation with zinc chloride and Negishi cross-coupling were unsuccessful, which does confirm that our deprotonation/carbometallation sequence is not suitable for the preparation of C-2-substituted (1,4-dihydro)pyridines.
![[1860-5397-8-250-i5]](/bjoc/content/inline/1860-5397-8-250-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: 2,3-Disubstituted pyridines by trapping of the intermediate metallated 1,4-dihydropyridine.
Scheme 5: 2,3-Disubstituted pyridines by trapping of the intermediate metallated 1,4-dihydropyridine.
Application to a formal synthesis of sarizotan
To further probe the synthetic utility of our pyridine synthesis, we next envisioned its use for the synthesis of the 3,5-disubstituted pyridine core of the antidyskinetic drug, 5-HT1A receptor agonist, dopamine D2 receptor ligand sarizotan 19 (Scheme 6). The pyridinyl-chroman sarizotan (also called EMD-128130) was originally developed by Merck KGaA in the late 1990’s [44] and was found to be a dual selective 5-HT1A receptor agonist and D2 receptor antagonist displaying a strong efficacy in the reduction of dyskinesia resulting from long-term antiparkinsonian treatment with levodopa [45-50]. Although its development was stopped by Merck KGaA in 2006 after analysis of data from Phase III clinical trials failed to confirm its efficiency [51], sarizotan is still under intense investigation [52-54] and was recently licensed to Newron Pharmaceuticals for further testing in new indications [55].
Motivated by the high potential of sarizotan and by the clear lack of structure-activity relationship studies on the pyridine core of this bioactive compound [56], we designed an efficient and modular synthesis of the disubstituted pyridine core of sarizotan that should enable the preparation of sarizotan analogues with different substitution on the pyridine ring. This synthesis is shown in Scheme 6 and starts from commercially available 2-methylene-1,3-propanediol (11). Mono-protection of this diol as a TBS ether and activation of the remaining alcohol as a mesylate according to previously reported procedures [57,58], gave allylic mesylate 12, which was next reacted with potassium phthalimide in DMF at 90 °C, affording the corresponding N-allylphthalimide. Deprotection of the phthalimide by hydrazinolysis followed by protection of the resulting primary amine as a tert-butyl-carbamate gave the Boc-protected amine 13 required for the synthesis of the substrate of the anionic cyclization. Indeed, this amine 13 was engaged in a copper-catalyzed cross-coupling with 1-(bromoethynyl)-4-fluorobenzene (14) and gave the corresponding ynamide 15 in a modest, unoptimized 36% yield. This set the stage for the key lithiation/intramolecular carbolithiation/oxidation step for the formation of the pyridine ring. To our delight, treatment of 14 under our optimized conditions (treatment with s-butyllithium and tetramethylethylenediamine in THF at −78 °C for one hour followed by addition of acetic acid and o-chloranil) smoothly promoted the anionic 6-endo-dig cyclization to the 3,5-disubstituted pyridine 16, which was isolated in 61% yield. Further deprotection of the primary alcohol with TBAF followed by chlorination with thionyl chloride finally gave the desired chloromethylpyridine 17, an intermediate used for the preparation of sarizotan 19 at Merck KGaA by coupling of this pyridine fragment 17 with aminomethylchroman 18 [44].
![[1860-5397-8-250-i6]](/bjoc/content/inline/1860-5397-8-250-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formal synthesis of the anti-dyskinesia agent, 5-HT1A receptor agonist, dopamine D2 receptor ligand sarizotan.
Scheme 6: Formal synthesis of the anti-dyskinesia agent, 5-HT1A receptor agonist, dopamine D2 receptor ligand...
Conclusion
The lithiation/isomerization/6-endo-dig intramolecular carbolithiation sequence from readily available N-allyl-ynamides provides an efficient entry to highly substituted 1,4-dihydropyridines and pyridines and has been successfully implemented in a formal synthesis of the anti-dyskinesia agent sarizotan. This new addition to the field of carbometallation reactions extends the chemistry of ynamides and should be useful in heterocyclic and medicinal chemistry as well. Further studies to extend this process to other heteroatom-substituted alkynes and to develop an asymmetric version of our 1,4-dihydropyridines synthesis are in progress and will be reported in due timecourse.
Acknowledgements
The authors thank the CNRS, the Universities of Versailles, Monastir and Brussels, the ANR (project DYNAMITE ANR-2010-BLAN-704) and the CMCU/PHC Utique (grant 10G1025) for support. We are grateful to Ms. Estelle Galmiche and Mr. Flavien Bourdreux (Lavoisier Institute) for their assistance with HRMS and NMR analyses and to Mr. Guillaume Boissonnat (Université Libre de Bruxelles and Ecole Polytechnique ParisTech) for technical assistance.
References
-
Ziegler, K.; Bähr, K. Ber. Dtsch. Chem. Ges. 1928, 253–263. doi:10.1002/cber.19280610203
Return to citation in text: [1] -
Clayden, J. Organolithium: Selectivity for Synthesis; Elsevier: Oxford, 2002.
Return to citation in text: [1] -
Marek, I.; Chinkov, N.; Banon-Tenne, D. Carbometallation Reactions. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F.; de Meijere, A., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 395–478.
Return to citation in text: [1] -
Fañanás, F. J.; Sanz, R. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; Patai series; Wiley: Chichester, 2006; pp 295–379.
Return to citation in text: [1] -
Chemla, F.; Ferreira, F. In The Chemistry of Organocopper Compounds; Rappoport, Z.; Marek, I., Eds.; Patai series; Wiley: Chichester, 2009; pp 527–584.
Return to citation in text: [1] -
Basheer, A.; Marek, I. Beilstein J. Org. Chem. 2010, 6, No. 77. doi:10.3762/bjoc.6.77
Return to citation in text: [1] -
Coste, A.; Karthikeyan, G.; Couty, F.; Evano, G. Angew. Chem., Int. Ed. 2009, 48, 4381–4385. doi:10.1002/anie.200901099
Return to citation in text: [1] [2] -
Jouvin, K.; Couty, F.; Evano, G. Org. Lett. 2010, 12, 3272–3275. doi:10.1021/ol101322k
Return to citation in text: [1] [2] -
Fadel, A.; Legrand, F.; Evano, G.; Rabasso, N. Adv. Synth. Catal. 2011, 353, 263–267. doi:10.1002/adsc.201000792
Return to citation in text: [1] -
Jouvin, K.; Evano, G. Chim. Oggi 2011, 29, 31–33.
Return to citation in text: [1] -
Laouiti, A.; Rammah, M. M.; Rammah, M. B.; Marrot, J.; Couty, F.; Evano, G. Org. Lett. 2012, 14, 6–9. doi:10.1021/ol2032152
Return to citation in text: [1] -
Jouvin, K.; Heimburger, J.; Evano, G. Chem. Sci. 2012, 3, 756–760. doi:10.1039/c2sc00842d
Return to citation in text: [1] [2] -
Laouiti, A.; Jouvin, K.; Bourdreux, F.; Rammah, M. M.; Rammah, M. B.; Evano, G. Synthesis 2012, 44, 1491–1500. doi:10.1055/s-0031-1289760
Return to citation in text: [1] -
Compain, G.; Jouvin, K.; Martin-Mingot, A.; Evano, G.; Marrot, J.; Thibaudeau, S. Chem. Commun. 2012, 48, 5196–5198. doi:10.1039/c2cc31768k
Return to citation in text: [1] -
Jouvin, K.; Coste, A.; Bayle, A.; Legrand, F.; Karthikeyan, G.; Tadiparthi, K.; Evano, G. Organometallics 2012, 31, 7933–7947.
Return to citation in text: [1] -
Evano, G.; Coste, A.; Jouvin, K. Angew. Chem., Int. Ed. 2010, 49, 2840–2859. doi:10.1002/anie.200905817
Return to citation in text: [1] -
DeKorver, K. A.; Li, H.; Lohse, A. G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R. P. Chem. Rev. 2010, 110, 5064–5106. doi:10.1021/cr100003s
Return to citation in text: [1] -
Evano, G.; Jouvin, K.; Coste, A. Synthesis 2013, 45, 17–26. doi:10.1055/s-0032-1317880.
Return to citation in text: [1] -
Couty, S.; Liégault, B.; Meyer, C.; Cossy, J. Org. Lett. 2004, 6, 2511–2514. doi:10.1021/ol049302m
Return to citation in text: [1] -
Couty, S.; Liegault, B.; Meyer, C.; Cossy, J. Tetrahedron 2006, 62, 3882–3895. doi:10.1016/j.tet.2005.11.089
Return to citation in text: [1] -
Couty, S.; Meyer, C.; Cossy, J. Tetrahedron Lett. 2006, 47, 767–769. doi:10.1016/j.tetlet.2005.11.093
Return to citation in text: [1] -
Chechik-Lankin, H.; Livshin, S.; Marek, I. Synlett 2005, 2098–2100. doi:10.1055/s-2005-871962
Return to citation in text: [1] -
Das, J. P.; Chechik, H.; Marek, I. Nat. Chem. 2009, 1, 128–132. doi:10.1038/nchem.131
Return to citation in text: [1] -
Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569
Return to citation in text: [1] -
Gourdet, B.; Lam, H. W. J. Am. Chem. Soc. 2009, 131, 3802–3803. doi:10.1021/ja900946h
Return to citation in text: [1] -
Gourdet, B.; Rudkin, M. E.; Watts, C. A.; Lam, H. W. J. Org. Chem. 2009, 74, 7849–7858. doi:10.1021/jo901658v
Return to citation in text: [1] -
Lavilla, R. J. Chem. Soc., Perkin Trans. 1 2002, 1141–1156. doi:10.1039/B101371H
Return to citation in text: [1] -
Moreau, J.; Hurvois, J.-P.; Mbaye, M. D.; Renaud, J.-L. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Springer: New York, 2009; Vol. 13, pp 201–230.
Return to citation in text: [1] -
Bull, J. A.; Mousseau, J. J.; Pelletier, G.; Charette, A. B. Chem. Rev. 2012, 112, 2642–2713. doi:10.1021/cr200251d
Return to citation in text: [1] -
Lukevitz, É. Chem. Heterocycl. Compd. 1995, 31, 639–650. doi:10.1007/BF01169065
Return to citation in text: [1] -
Jones, G. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V.; McKillop, A., Eds.; Pergamon: Oxford, 1996; Vol. 5, pp 167–243.
Return to citation in text: [1] -
Henry, G. D. Tetrahedron 2004, 60, 6043–6061. doi:10.1016/j.tet.2004.04.043
Return to citation in text: [1] -
Spitzner, D. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Black, D., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 15, pp 11–285.
Return to citation in text: [1] -
Park, Y. S.; Weisenburger, G. A.; Beak, P. J. Am. Chem. Soc. 1997, 119, 10537–10538. doi:10.1021/ja972333a
Return to citation in text: [1] -
Curtis, M. D.; Beak, P. J. Org. Chem. 1999, 64, 2996–2997. doi:10.1021/jo990383n
Return to citation in text: [1] -
Lim, S. H.; Curtis, M. D.; Beak, P. Org. Lett. 2001, 3, 711–714. doi:10.1021/ol007012+
Return to citation in text: [1] -
Johnson, T. A.; Jang, D. O.; Slafer, B. W.; Curtis, M. D.; Beak, P. J. Am. Chem. Soc. 2002, 124, 11689–11698. doi:10.1021/ja0271375
Return to citation in text: [1] -
Beak, P.; Lee, B. J. Org. Chem. 1989, 54, 458–464. doi:10.1021/jo00263a037
Return to citation in text: [1] -
Gilmore, K.; Alabugin, I. V. Chem. Rev. 2011, 111, 6513–6556. doi:10.1021/cr200164y
Return to citation in text: [1] -
Gati, W.; Rammah, M. M.; Rammah, M. B.; Couty, F.; Evano, F. J. Am. Chem. Soc. 2012, 134, 9078–9081. doi:10.1021/ja303002a
Return to citation in text: [1] -
Comins, D. L. Tetrahedron Lett. 1983, 24, 2807–2810. doi:10.1016/S0040-4039(00)88029-6
Return to citation in text: [1] -
Zhang, Y.; Hsung, R. P.; Tracey, M. R.; Kurtz, K. C. M.; Vera, E. L. Org. Lett. 2004, 6, 1151–1154. doi:10.1021/ol049827e
Return to citation in text: [1] -
Hamada, T.; Ye, X.; Stahl, S. S. J. Am. Chem. Soc. 2008, 130, 833–835. doi:10.1021/ja077406x
Return to citation in text: [1] -
Bottcher, H.; DeVant, R.; Greiner, H.; Bartoszyk, G.; Berthelon, J.-J.; Noblet, M.; Zeiller, J.-J.; Brunet, M. Pyridyl chroman. U.S. patent 5,767,132, June 16, 1998.
Return to citation in text: [1] [2] -
Bibbiani, F.; Oh, J. D.; Chase, T. N. Neurology 2001, 57, 1829–1834. doi:10.1212/WNL.57.10.1829
Return to citation in text: [1] -
Rabiner, E. A.; Gunn, R. N.; Wilkins, M. R.; Sedman, E.; Grasby, P. M. J. Psychopharmacol. (London, U. K.) 2002, 16, 195–199. doi:10.1177/026988110201600301
Return to citation in text: [1] -
Bartoszyk, G.; Van Amsterdam, C.; Greiner, H.; Rautenberg, W.; Russ, H.; Seyfried, C. J. Neural. Transm. 2004, 111, 113–126. doi:10.1007/s00702-003-0094-7
Return to citation in text: [1] -
Olanow, C. W.; Damier, P.; Goetz, C. G.; Mueller, T.; Nutt, J.; Rascol, O.; Serbanescu, A.; Deckers, F.; Russ, H. Clin. Neuropharmacol. 2004, 27, 58–62. doi:10.1097/00002826-200403000-00003
Return to citation in text: [1] -
Bara-Jimenez, W.; Bibbiani, F.; Morris, M.; Dimitrova, T.; Sherzai, A.; Mouradian, M.; Chase, T. Movement Disord. 2005, 20, 932–936. doi:10.1002/mds.20370
Return to citation in text: [1] -
Grégoire, L.; Samadi, P.; Graham, J.; Bédard, P.; Bartoszyk, G.; Di Paolo, T. Parkinsonism Relat. Disord. 2009, 15, 445–452. doi:10.1016/j.parkreldis.2008.11.001
Return to citation in text: [1] -
Merck news release. http://me.merck.de/n/A720D2336B86597DC1257196001CCA99/$FILE/Sarizo-e.pdf (accessed Dec 1, 2012).
Return to citation in text: [1] -
Gallemann, D.; Wimmer, E.; Hofer, C. C.; Freisleben, A.; Fluck, M.; Ladstetter, B.; Dolgos, H. Drug Metab. Dispos. 2010, 38, 905–916. doi:10.1124/dmd.109.029835
Return to citation in text: [1] -
Zhang, X. Q.; Egeland, M.; Svenningsson, P. Psychopharmacology 2011, 218, 621–634. doi:10.1007/s00213-011-2356-7
Return to citation in text: [1] -
Gerlach, M.; Bartoszyk, G. D.; Riederer, P.; Dean, O.; van den Buuse, M. J. Neural Transm. 2011, 118, 1733–1742. doi:10.1007/s00702-010-0571-8
Return to citation in text: [1] -
Molecules under development at Newron Pharmaceuticals. http://www.newron.com/Pipeline.html (accessed Dec 1, 2012).
Return to citation in text: [1] -
A single analogue possessing a 3,4-difluorophenyl ring in place of the 4-fluorophenyl has been reported in ref. [44], while extensive modifications have been made on the chroman core.
Return to citation in text: [1] -
Fadayi, O. O.; Senter, T. J.; Hahn, K. N.; Lindsley, C. W. Chem.–Eur. J. 2012, 18, 5826–5831. doi:10.1002/chem.201200629
Return to citation in text: [1] -
Couladourous, E. A.; Dakanali, M.; Demadis, K. D.; Vidali, V. P. Org. Lett. 2009, 11, 4430–4433. doi:10.1021/ol901781n
Return to citation in text: [1]
| 1. | Ziegler, K.; Bähr, K. Ber. Dtsch. Chem. Ges. 1928, 253–263. doi:10.1002/cber.19280610203 |
| 19. | Couty, S.; Liégault, B.; Meyer, C.; Cossy, J. Org. Lett. 2004, 6, 2511–2514. doi:10.1021/ol049302m |
| 20. | Couty, S.; Liegault, B.; Meyer, C.; Cossy, J. Tetrahedron 2006, 62, 3882–3895. doi:10.1016/j.tet.2005.11.089 |
| 21. | Couty, S.; Meyer, C.; Cossy, J. Tetrahedron Lett. 2006, 47, 767–769. doi:10.1016/j.tetlet.2005.11.093 |
| 43. | Hamada, T.; Ye, X.; Stahl, S. S. J. Am. Chem. Soc. 2008, 130, 833–835. doi:10.1021/ja077406x |
| 7. | Coste, A.; Karthikeyan, G.; Couty, F.; Evano, G. Angew. Chem., Int. Ed. 2009, 48, 4381–4385. doi:10.1002/anie.200901099 |
| 8. | Jouvin, K.; Couty, F.; Evano, G. Org. Lett. 2010, 12, 3272–3275. doi:10.1021/ol101322k |
| 9. | Fadel, A.; Legrand, F.; Evano, G.; Rabasso, N. Adv. Synth. Catal. 2011, 353, 263–267. doi:10.1002/adsc.201000792 |
| 10. | Jouvin, K.; Evano, G. Chim. Oggi 2011, 29, 31–33. |
| 11. | Laouiti, A.; Rammah, M. M.; Rammah, M. B.; Marrot, J.; Couty, F.; Evano, G. Org. Lett. 2012, 14, 6–9. doi:10.1021/ol2032152 |
| 12. | Jouvin, K.; Heimburger, J.; Evano, G. Chem. Sci. 2012, 3, 756–760. doi:10.1039/c2sc00842d |
| 13. | Laouiti, A.; Jouvin, K.; Bourdreux, F.; Rammah, M. M.; Rammah, M. B.; Evano, G. Synthesis 2012, 44, 1491–1500. doi:10.1055/s-0031-1289760 |
| 14. | Compain, G.; Jouvin, K.; Martin-Mingot, A.; Evano, G.; Marrot, J.; Thibaudeau, S. Chem. Commun. 2012, 48, 5196–5198. doi:10.1039/c2cc31768k |
| 15. | Jouvin, K.; Coste, A.; Bayle, A.; Legrand, F.; Karthikeyan, G.; Tadiparthi, K.; Evano, G. Organometallics 2012, 31, 7933–7947. |
| 16. | Evano, G.; Coste, A.; Jouvin, K. Angew. Chem., Int. Ed. 2010, 49, 2840–2859. doi:10.1002/anie.200905817 |
| 17. | DeKorver, K. A.; Li, H.; Lohse, A. G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R. P. Chem. Rev. 2010, 110, 5064–5106. doi:10.1021/cr100003s |
| 18. | Evano, G.; Jouvin, K.; Coste, A. Synthesis 2013, 45, 17–26. doi:10.1055/s-0032-1317880. |
| 7. | Coste, A.; Karthikeyan, G.; Couty, F.; Evano, G. Angew. Chem., Int. Ed. 2009, 48, 4381–4385. doi:10.1002/anie.200901099 |
| 6. | Basheer, A.; Marek, I. Beilstein J. Org. Chem. 2010, 6, No. 77. doi:10.3762/bjoc.6.77 |
| 41. | Comins, D. L. Tetrahedron Lett. 1983, 24, 2807–2810. doi:10.1016/S0040-4039(00)88029-6 |
| 2. | Clayden, J. Organolithium: Selectivity for Synthesis; Elsevier: Oxford, 2002. |
| 3. | Marek, I.; Chinkov, N.; Banon-Tenne, D. Carbometallation Reactions. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F.; de Meijere, A., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 395–478. |
| 4. | Fañanás, F. J.; Sanz, R. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; Patai series; Wiley: Chichester, 2006; pp 295–379. |
| 5. | Chemla, F.; Ferreira, F. In The Chemistry of Organocopper Compounds; Rappoport, Z.; Marek, I., Eds.; Patai series; Wiley: Chichester, 2009; pp 527–584. |
| 42. | Zhang, Y.; Hsung, R. P.; Tracey, M. R.; Kurtz, K. C. M.; Vera, E. L. Org. Lett. 2004, 6, 1151–1154. doi:10.1021/ol049827e |
| 30. | Lukevitz, É. Chem. Heterocycl. Compd. 1995, 31, 639–650. doi:10.1007/BF01169065 |
| 31. | Jones, G. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V.; McKillop, A., Eds.; Pergamon: Oxford, 1996; Vol. 5, pp 167–243. |
| 32. | Henry, G. D. Tetrahedron 2004, 60, 6043–6061. doi:10.1016/j.tet.2004.04.043 |
| 33. | Spitzner, D. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Black, D., Ed.; Thieme: Stuttgart, Germany, 2004; Vol. 15, pp 11–285. |
| 39. | Gilmore, K.; Alabugin, I. V. Chem. Rev. 2011, 111, 6513–6556. doi:10.1021/cr200164y |
| 27. | Lavilla, R. J. Chem. Soc., Perkin Trans. 1 2002, 1141–1156. doi:10.1039/B101371H |
| 28. | Moreau, J.; Hurvois, J.-P.; Mbaye, M. D.; Renaud, J.-L. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Springer: New York, 2009; Vol. 13, pp 201–230. |
| 29. | Bull, J. A.; Mousseau, J. J.; Pelletier, G.; Charette, A. B. Chem. Rev. 2012, 112, 2642–2713. doi:10.1021/cr200251d |
| 40. | Gati, W.; Rammah, M. M.; Rammah, M. B.; Couty, F.; Evano, F. J. Am. Chem. Soc. 2012, 134, 9078–9081. doi:10.1021/ja303002a |
| 25. | Gourdet, B.; Lam, H. W. J. Am. Chem. Soc. 2009, 131, 3802–3803. doi:10.1021/ja900946h |
| 26. | Gourdet, B.; Rudkin, M. E.; Watts, C. A.; Lam, H. W. J. Org. Chem. 2009, 74, 7849–7858. doi:10.1021/jo901658v |
| 22. | Chechik-Lankin, H.; Livshin, S.; Marek, I. Synlett 2005, 2098–2100. doi:10.1055/s-2005-871962 |
| 23. | Das, J. P.; Chechik, H.; Marek, I. Nat. Chem. 2009, 1, 128–132. doi:10.1038/nchem.131 |
| 24. | Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569 |
| 34. | Park, Y. S.; Weisenburger, G. A.; Beak, P. J. Am. Chem. Soc. 1997, 119, 10537–10538. doi:10.1021/ja972333a |
| 35. | Curtis, M. D.; Beak, P. J. Org. Chem. 1999, 64, 2996–2997. doi:10.1021/jo990383n |
| 36. | Lim, S. H.; Curtis, M. D.; Beak, P. Org. Lett. 2001, 3, 711–714. doi:10.1021/ol007012+ |
| 37. | Johnson, T. A.; Jang, D. O.; Slafer, B. W.; Curtis, M. D.; Beak, P. J. Am. Chem. Soc. 2002, 124, 11689–11698. doi:10.1021/ja0271375 |
| 38. | Beak, P.; Lee, B. J. Org. Chem. 1989, 54, 458–464. doi:10.1021/jo00263a037 |
| 44. | Bottcher, H.; DeVant, R.; Greiner, H.; Bartoszyk, G.; Berthelon, J.-J.; Noblet, M.; Zeiller, J.-J.; Brunet, M. Pyridyl chroman. U.S. patent 5,767,132, June 16, 1998. |
| 8. | Jouvin, K.; Couty, F.; Evano, G. Org. Lett. 2010, 12, 3272–3275. doi:10.1021/ol101322k |
| 12. | Jouvin, K.; Heimburger, J.; Evano, G. Chem. Sci. 2012, 3, 756–760. doi:10.1039/c2sc00842d |
| 44. | Bottcher, H.; DeVant, R.; Greiner, H.; Bartoszyk, G.; Berthelon, J.-J.; Noblet, M.; Zeiller, J.-J.; Brunet, M. Pyridyl chroman. U.S. patent 5,767,132, June 16, 1998. |
| 44. | Bottcher, H.; DeVant, R.; Greiner, H.; Bartoszyk, G.; Berthelon, J.-J.; Noblet, M.; Zeiller, J.-J.; Brunet, M. Pyridyl chroman. U.S. patent 5,767,132, June 16, 1998. |
| 56. | A single analogue possessing a 3,4-difluorophenyl ring in place of the 4-fluorophenyl has been reported in ref. [44], while extensive modifications have been made on the chroman core. |
| 57. | Fadayi, O. O.; Senter, T. J.; Hahn, K. N.; Lindsley, C. W. Chem.–Eur. J. 2012, 18, 5826–5831. doi:10.1002/chem.201200629 |
| 58. | Couladourous, E. A.; Dakanali, M.; Demadis, K. D.; Vidali, V. P. Org. Lett. 2009, 11, 4430–4433. doi:10.1021/ol901781n |
| 52. | Gallemann, D.; Wimmer, E.; Hofer, C. C.; Freisleben, A.; Fluck, M.; Ladstetter, B.; Dolgos, H. Drug Metab. Dispos. 2010, 38, 905–916. doi:10.1124/dmd.109.029835 |
| 53. | Zhang, X. Q.; Egeland, M.; Svenningsson, P. Psychopharmacology 2011, 218, 621–634. doi:10.1007/s00213-011-2356-7 |
| 54. | Gerlach, M.; Bartoszyk, G. D.; Riederer, P.; Dean, O.; van den Buuse, M. J. Neural Transm. 2011, 118, 1733–1742. doi:10.1007/s00702-010-0571-8 |
| 55. | Molecules under development at Newron Pharmaceuticals. http://www.newron.com/Pipeline.html (accessed Dec 1, 2012). |
| 45. | Bibbiani, F.; Oh, J. D.; Chase, T. N. Neurology 2001, 57, 1829–1834. doi:10.1212/WNL.57.10.1829 |
| 46. | Rabiner, E. A.; Gunn, R. N.; Wilkins, M. R.; Sedman, E.; Grasby, P. M. J. Psychopharmacol. (London, U. K.) 2002, 16, 195–199. doi:10.1177/026988110201600301 |
| 47. | Bartoszyk, G.; Van Amsterdam, C.; Greiner, H.; Rautenberg, W.; Russ, H.; Seyfried, C. J. Neural. Transm. 2004, 111, 113–126. doi:10.1007/s00702-003-0094-7 |
| 48. | Olanow, C. W.; Damier, P.; Goetz, C. G.; Mueller, T.; Nutt, J.; Rascol, O.; Serbanescu, A.; Deckers, F.; Russ, H. Clin. Neuropharmacol. 2004, 27, 58–62. doi:10.1097/00002826-200403000-00003 |
| 49. | Bara-Jimenez, W.; Bibbiani, F.; Morris, M.; Dimitrova, T.; Sherzai, A.; Mouradian, M.; Chase, T. Movement Disord. 2005, 20, 932–936. doi:10.1002/mds.20370 |
| 50. | Grégoire, L.; Samadi, P.; Graham, J.; Bédard, P.; Bartoszyk, G.; Di Paolo, T. Parkinsonism Relat. Disord. 2009, 15, 445–452. doi:10.1016/j.parkreldis.2008.11.001 |
| 51. | Merck news release. http://me.merck.de/n/A720D2336B86597DC1257196001CCA99/$FILE/Sarizo-e.pdf (accessed Dec 1, 2012). |
© 2012 Gati et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)