Abstract
The synthetic protocol for the reduction of alcohols to hydrocarbons by using hydriodic acid, first described by Kiliani more than 140 years ago, was improved to be more applicable to organic synthesis. Instead of a strongly acidic, aqueous solution, a biphasic toluene–water reaction medium was used, which allowed the conversion of primary, secondary and tertiary benzylic alcohols, in good yields and short reaction times, into the corresponding hydrocarbons. Red phosphorous was used as the stoichiometric reducing agent. Keto, ester, amide or ether groups are tolerated, and catalytic amounts of hydriodic acid (0.2 equiv) in the presence of 0.6 equiv phosphorous are sufficient to achieve conversion.

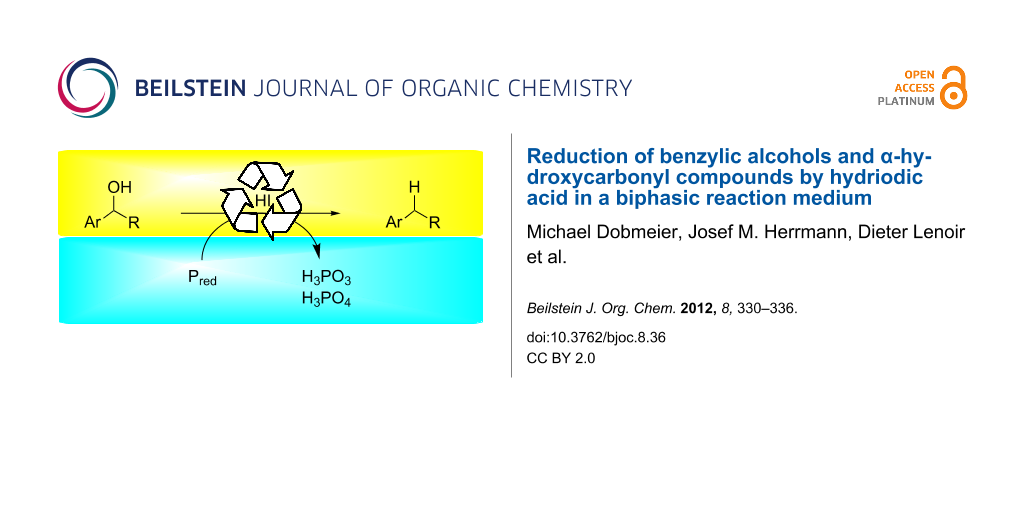
Graphical Abstract
Introduction
The reduction of hydroxy groups is a typical and important step in the synthesis of complex natural products or drugs [1-4]. Functional-group tolerance during this reduction step is essential since various other groups are usually present. A number of synthetic procedures have been developed, which allow selective reduction, but only a few one-step transformations are known, which use either titanium(III) [5-8] or different metal complexes [9-13]. Most procedures require a sequence of steps, e.g., the conversion of hydroxy groups into a chloride or bromide substituent and subsequent catalytic reduction with H2/Pt or the conversion into a tosylate and reduction with LiAlH4. The most commonly applied method is the Barton–McCombie reaction [14], due to its versatility and its very high functional-group tolerance [15-18]. Although very general, the reaction has some drawbacks: The involved organotin hydrides are costly, highly toxic [19-21] and often difficult to separate from the reaction products. Furthermore, secondary alcohols give best results, while others may react less efficiently.
We have reinvestigated the long-known reduction of benzylic alcohols and α-hydroxycarbonyl compounds by hydriodic acid [22-32]. The method has been reported for a variety of alcohols, but typically proceeds in aqueous solution and requires an excess of HI or strong mineral acids such as phosphoric or sulfuric acid [33-35]. We describe a biphasic reaction medium consisting of toluene and aqueous hydriodic acid. The phase separation allows milder reaction conditions compared to the classic Kiliani protocol and is more applicable to organic synthesis.
Results and Discussion
Initial investigations focused on simple benzylic alcohols (Table 1, entries 1–3), which were converted in high to quantitative yields into the corresponding alkanes. Carbonyl groups or amides in a benzylic position (Table 1, entries 4 and 6) and aromatic hydroxy groups (Table 2, entry 7) or aromatic ethers (Table 1, entry 5) were not affected. Moreover, heterocycles such as thiophene (Table 1, entry 7) were stable under these conditions whereas furans (Table 1, entry 8) were decomposed due to ring opening. Benzylic alcohols were converted in good to high yields to alkanes with increasing reactivity in the order primary (2 h) < secondary (0.5–1 h) < tertiary alcohol (15–30 min); carbonyl groups and ethers were tolerated. Diethyl tartrate was converted into diethyl succinate under the reaction conditions given (Table 1, entry 12), but some of the material was lost due to ester hydrolysis.
Table 1: Reduction of benzylic alcohols to the corresponding alkanes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-36-i2.svg?max-width=637&scale=1.0)
|
||||
| Entry | Alcohol | Producta | Time [h] | Yield [%] |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-36-i3.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-36-i4.svg?max-width=637&scale=1.0)
|
2 | 70b |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-36-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-36-i6.svg?max-width=637&scale=1.0)
|
0.5 | 96 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-36-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-36-i8.svg?max-width=637&scale=1.0)
|
0.25 | 100 |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-36-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-36-i10.svg?max-width=637&scale=1.0)
|
1 | 80 |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-36-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-36-i12.svg?max-width=637&scale=1.0)
|
0.5 | 92 |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-36-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-36-i14.svg?max-width=637&scale=1.0)
|
1 | 82 |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-36-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-36-i16.svg?max-width=637&scale=1.0)
|
0.5 | 62c |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-36-i17.svg?max-width=637&scale=1.0)
|
decomposition | 1 | – |
| 9 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-36-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-36-i19.svg?max-width=637&scale=1.0)
|
0.25 | 74c |
| 10 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-36-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-36-i21.svg?max-width=637&scale=1.0)
|
0.5 | 49c |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-36-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-36-i23.svg?max-width=637&scale=1.0)
|
0.5 | 78 |
| 12 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-36-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-36-i25.svg?max-width=637&scale=1.0)
|
1.5 | 65 |
aAll products are known compounds described in the literature. The identities have been proven by proton NMR and mass analysis, which match the literature data. bThe corresponding iodo compound was identified as a byproduct. cThe corresponding elimination product was obtained as a byproduct.
Table 2: Alcohols showing incomplete or unselective reaction with hydriodic acid and red phosphorous (3.0 equiv HI, 0.4 equiv Pred).
| Entry | Alcohol | Product | Time [h] | Yield [%] |
|---|---|---|---|---|
| 1 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-36-i26.svg?max-width=637&scale=1.0)
|
mixture of several products | 1 | – |
| 2 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-36-i27.svg?max-width=637&scale=1.0)
|
mixture of several products | 1 | – |
| 3 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-36-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-8-36-i29.svg?max-width=637&scale=1.0)
|
1 | traces |
| 4 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-8-36-i30.svg?max-width=637&scale=1.0)
|
decomposition | 1 | – |
| 5 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-8-36-i31.svg?max-width=637&scale=1.0)
|
decomposition | 1 | – |
| 6 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-8-36-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-8-36-i33.svg?max-width=637&scale=1.0)
|
2 | 21 |
| 7 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-8-36-i34.svg?max-width=637&scale=1.0)
|
no conversion | 1 | – |
Allylic alcohols are completely consumed, but the corresponding alkenes could not be isolated as pure products (Table 2). Mixtures of eliminiation and deoxygenation products, in some cases also rearangement of the deoxygenated product into the the more highly substitued, thermodynamically more stable alkene occurred. Propargylic alcohols (Table 2, entry 3 and 4) showed elimination or decomposed. In the case of flavin (Table 2, entry 6), three hydroxy groups were reduced and one was converted into an iodo substituent.
Alcohols other than those that were benzylic or α to carbonyl groups were not converted into the corresponding alkanes, and the reaction stopped at the iodoalkanes (Table 3). The reactivity follows the order of primary < secondary < tertiary alcohols, as expected for an SN1 reaction. The reduction potential of the nonbenzylic iodoalkanes is not sufficient for reduction by hydriodic acid.
Table 3: Alcohols yielding alkyl iodides with hydriodic acid and red phosphorous.a
| Entry | Alcohol | Product | Time [h] | Yield [%] |
|---|---|---|---|---|
| 1 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-8-36-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 35]](/bjoc/content/inline/1860-5397-8-36-i36.svg?max-width=637&scale=1.0)
|
8 | 98 |
| 2 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-8-36-i37.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-8-36-i38.svg?max-width=637&scale=1.0)
|
8 | 83b |
| 3 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-8-36-i39.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-8-36-i40.svg?max-width=637&scale=1.0)
|
20 | 81c |
a3 equiv HI, 0.4 equiv Pred. bSingle isomer. cProducts were analyzed by gas chromatography; chlorobenzene was used as an internal standard.
The mechanism of reduction by hydriodic acid consists of two steps (Scheme 1): The nucleophilic substitution of the hydroxy group by iodide and the subsequent reduction of the alkyl iodide by hydriodic acid. The mechanistic details of the redox comproportionation of alkyl iodides and H–I have been strongly debated in the literature [36-38]. However, the required benzylic or α-carbonyl position for the redox comproportionation indicates an intermediate with mesomeric stabilization due to the adjacent π-system. In a trapping experiment, using HI without phosphorous, diphenylcarbinol as the substrate and TEMPO as a trapping agent for radical intermediates, the TEMPO adduct of diphenylcarbinol was detected by mass analysis. This indicates a radical mechanism of the redox comproportionation. We suggest a stepwise reduction by single electron transfer (SET) accompanied by the oxidation of I− to I2. The iodine, generated in the second step, is recycled by reduction with red phosphorous, regenerating hydriodic acid. Admittedly, the above-mentioned TEMPO adduct could also be generated by nucleophilic substitution of the alkyl iodide with reduced TEMPO. At least this would be another proof for the first reaction step. According to the redox equations of the reaction between iodine and red phosphorous, each mole of red phosphorous is able to reduce at least 1.5 mol of iodine. Catalytic amounts of hydriodic acid are therefore sufficient [28] for the reduction of the hydroxy group (Table 4), when excess red phosphorous is added as a terminal reducing agent. However, depending on the amount of added hydriodic acid, the elimination of water may occur as an alternative reaction pathway. A low concentration of HI favors the elimination of water, while higher HI concentrations lead to substitution and reduction products.
![[1860-5397-8-36-i1]](/bjoc/content/inline/1860-5397-8-36-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanism of the alcohol reduction and recycling of iodine.
Scheme 1: Mechanism of the alcohol reduction and recycling of iodine.
![[Graphic 40]](/bjoc/content/inline/1860-5397-8-36-i41.svg?max-width=637&scale=1.0)
![[Graphic 41]](/bjoc/content/inline/1860-5397-8-36-i42.svg?max-width=637&scale=1.0)
![[Graphic 42]](/bjoc/content/inline/1860-5397-8-36-i43.svg?max-width=637&scale=1.0)
![[Graphic 43]](/bjoc/content/inline/1860-5397-8-36-i44.svg?max-width=637&scale=1.0)
![[Graphic 44]](/bjoc/content/inline/1860-5397-8-36-i45.svg?max-width=637&scale=1.0)
![[Graphic 45]](/bjoc/content/inline/1860-5397-8-36-i46.svg?max-width=637&scale=1.0)
![[Graphic 46]](/bjoc/content/inline/1860-5397-8-36-i47.svg?max-width=637&scale=1.0)
![[Graphic 47]](/bjoc/content/inline/1860-5397-8-36-i48.svg?max-width=637&scale=1.0)
Conclusion
Toluene and aqueous hydriodic acid are a suitable biphasic reaction mixture for the reduction of a range of benzylic alcohols. The two-phase system makes the Kiliani protocol more easily applicable to organic synthesis, as organic substrates and products dissolve in the organic phase and are separated from the mineral acids. The procedure allows the use of catalytic amounts of hydriodic acid and red phosphorous as the terminal reductant. In the case of alcohols having no activation by adjacent benzylic or carbonyl groups the reaction stops at the corresponding alkyl iodide. A quantitative mass-efficiency analysis [39] of the reaction in comparison to tosylation/LAH, Ti(III)-mediated and Barton–McCombie reduction revealed a better atom economy and mass efficiency.
Experimental
Representative experimental procedure: The alcohol (1 mmol, 1 equiv) is dissolved in 4 mL of toluene. Red phosphorus (0.4 mmol), followed by concentrated hydriodic acid (57% w/w; 3.0 mmol, 3 equiv) is added and the reaction mixture is heated to 80 °C for the stated time, allowed to cool to rt and quenched with Na2S2O3 (10 mL; 10% w/w) solution. The aqueous phase is extracted with dichloromethane (3 × 10 mL), the combined organic phases are dried over MgSO4 and filtered, and the solvent is removed. The crude product is purified by chromatography and spectroscopically characterized.
For catalytic reactions of 1 mmol of the respective alcohol the following amounts of hydriodic acid and Pred were used: (a) 0.6 mmol HI/0.4 mmol Pred, (b) 0.1 mmol HI/0.7 mmol Pred.
(E)-6-Methyl-1-phenylhept-4-en-3-ol (Table 2, entry 1): The reaction was carried out under dry nitrogen atmosphere by using standard Schlenk techniques. To a slurry of Mg powder (0.67 g, 28 mmol) in dry THF (4 mL), 2 mL of a solution of 2-phenyl-1-bromethane (3.0 mL, 28 mmol) in dry THF (10 mL) was added. The Grignard reaction was initiated by the addition of iodine followed by sonication for several minutes. When the exothermic reaction started the rest of the 2-phenyl-1-bromethane solution was added through a septum by syringe over 15 min. After the addition, the reaction solution was heated under reflux for 1 h to complete the reaction. The reaction solution was allowed to cool to rt before 4-methyl-2-pentenal (2.3 mL, 20 mmol) was added dropwise. To complete the reaction the solution was again heated under reflux for 1 h. The reaction was quenched by the addition of HCl (2 M, 25 mL). The aqueous phase was extracted with diethyl ether (3 × 15 mL). The combined organic phases were washed with saturated NaHCO3 (15 mL) and H2O (2 × 10 mL), and dried with MgSO4. The solvent was removed with a rotary evaporator. The crude product was purified by flash chromatography (petroleum ether/ethyl acetate 4:1, Rf 0.32; staining with vanillin solution gave a blue spot). (E)-6-Methyl-1-phenylhept-4-en-3-ol was isolated as a yellow oil in 74% yield (3.05 g, 14.9 mmol). 1H NMR (300 MHz, CDCl3) δ 7.33–7.14 (m, 5H), 5.63 (ddd, J = 15.5, 6.4, 0.7 Hz, 1H), 5.44 (ddd, J = 15.5, 7.0, 1.2 Hz, 1H), 4.13–4.01 (m, 1H), 2.79–2.59 (m, 2H), 2.39–2.21 (m, 1H), 1.97–1.72 (m, 2H), 1.58 (d, J = 2.7 Hz, 0.3H), 1.46 (d, J = 1.8 Hz, 1H), 1.00 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 139.6, 129.7, 128.5, 128.4, 125.8, 72.6, 38.8, 31.8, 30.7, 22.4, 21.3; EIMS m/z (%): 91.1 (100) [C7H7]+, 161.1 (81) [M − C3H7]+, 186.1 (5) [M − H2O]+, 204.2 [M]+∙; HRMS (m/z): [M]+ calcd for C14H20O, 204.1514; found, 204.1511.
(E)-1-Phenylhex-4-en-3-ol (Table 2, entry 2): The reaction was carried out under a dry nitrogen atmosphere by using standard Schlenk techniques. A solution (1 mL) of 2-phenyl-1-bromethane (1.35 mL, 10.0 mmol) in dry THF (10 mL) was added to Mg powder (0.25 g, 10 mmol). The Grignard reaction was initiated by the addition of iodine followed by sonication for several min. When the exothermic reaction started the rest of the 2-phenyl-1-bromethane solution was added through a septum by syringe over 15 min. After the addition, the reaction solution was heated under reflux for 1 h to complete the reaction. The reaction solution was allowed to cool to rt before crotonaldehyde (0.74 mL, 9.0 mmol) was added dropwise. To complete the reaction the solution was again heated under reflux for 2.5 h. The reaction was quenched by the addition of HCl (2 M, 10 mL). The aqueous phase was extracted with diethyl ether (2 × 15 mL). The combined organic phases were washed with saturated NaHCO3 (5 mL), H2O (2 × 5 mL) and dried with MgSO4. The solvent was removed with a rotary evaporator. (E)-1-Phenylhex-4-en-3-ol was obtained in 96% yield (1.53 g, 8.69 mmol) in analytical purity. Analytical data were identical with the literature [40]. 1H NMR (300 MHz, CDCl3) δ 7.34–7.06 (m, 5H), 5.63 (dq, J = 15.3, 6.2 Hz, 1H), 5.48 (ddd, J = 15.3, 7.0, 1.4 Hz, 1H), 4.02 (q, J = 6.7 Hz, 1H), 2.73–2.56 (m, 2H), 1.67 (dd, J = 6.3, 0.7 Hz, 3H), 1.52 (s, 0.3H), 1.40 (s, 0.7H); EIMS m/z (%): 71.1 (100) [C4H7O]+, 91.1 (67) [C7H7]+, 105.1 (19) [M − C4H7O]+, 176.1 (50) [M]+.
1-(4-Methoxyphenyl)-2-phenylpropan-1-ol (Table 1, entry 5): The reaction was carried out under a dry nitrogen atmosphere by using standard Schlenk techniques. 1 mL of a solution of 4-bromo-1-methoxybenzene (0.62 mL, 5.0 mmol) in dry THF (10 mL) was added to Mg powder (0.12 g, 5.0 mmol). The Grignard reaction was initiated by the addition of iodine followed by sonication for several min. When the exothermic reaction started the rest of the 4-bromo-1-methoxybenzene solution was added through a septum by syringe over 15 min. After the addition, the reaction solution was heated under reflux for 1 h to complete the reaction. The reaction solution was allowed to cool to rt before 2-phenylpropionaldehyde (0.60 mL, 4.5 mmol) was added dropwise. To complete the reaction the solution was again heated under reflux for 2 h. The reaction was quenched by the addition of HCl (2 M, 5 mL). The aqueous phase was extracted with diethyl ether (2 × 5 mL). The combined organic phases were washed with saturated NaHCO3 (3 mL), H2O (2 × 2.5 mL) and dried with MgSO4. The solvent was removed with a rotary evaporator. The crude product was purified by flash chromatography (petroleum ether/ethyl acetate 4:1, Rf 0.3; staining with vanillin solution gave a blue spot). 1-(4-Methoxyphenyl)-2-phenylpropan-1-ol was isolated as a yellow oil in 57% yield (0.62 g, 2.6 mmol). Analytical data are identical with literature [41]. 1H NMR (300 MHz, CDCl3) δ 7.45–7.05 (m, 7H), 6.85–6.74 (m, 2H), 4.76 (d, J = 6.1 Hz, 1H), 3.78 (s, 3H), 3.09 (p, J = 6.9 Hz, 1H), 1.34 (d, J = 7.0 Hz, 3H); EIMS m/z (%): 137.1 (53) [M − C8H9]+, 224.1 (2) [M − H2O]+, 242.1 (1) [M]+∙.
6,6-Dimethyl-2-phenylhept-4-yn-3-ol (Table 2, entry 4): The reaction was carried out under a dry nitrogen atmosphere by using standard Schlenk techniques. The solution of 3,3-dimethyl-1-butyne (0.62 mL, 5 mmol) in dry THF (10 mL) was cooled to −78 °C. n-BuLi (1.6 M in hexane, 3.5 mL, 5.6 mmol) was added dropwise through a septum by syringe. The reaction mixture was allowed to warm to rt before the solution of 2-propionaldehyde (0.68 mL, 5 mmol) in dry THF (5 mL) was added dropwise through a septum by syringe. This solution was stirred for 4.5 h. The reaction was stopped by the addition of H2O (10 mL). The aqueous phase was extracted with diethyl ether (3 × 15 mL), and the combined organic layers were dried with MgSO4. The solvent was removed with a rotary evaporator. The crude product was purified by flash chromatography (petroleum ether/ethyl acetate 4:1, Rf 0.42; staining with vanillin solution gave a blue spot). 6,6-dimethyl-2-phenylhept-4-yn-3-ol was isolated as a colorless oil in 46% yield (0.50 g, 2.3 mmol). 1H NMR (300 MHz, CDCl3) δ 7.40–7.19 (m, 5H), 4.44 (dd, J = 7.4, 5.4 Hz, 1H), 3.03 (dd, J = 7.1, 5.4 Hz, 1H), 1.67 (d, J = 5.4 Hz, 1H), 1.64 (d, J = 7.4 Hz, 1H), 1.39 (d, J = 7.1 Hz, 3H), 1.17 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 141.9, 128.8, 128.2, 127.0, 95.5, 78.1, 67.8, 67.5, 55.0, 46.1, 31.0, 30.0, 16.3; EIMS m/z (%): 57.1 (36) [C4H9]+, 99.1 (100), 105.1 (20) [C8H10]+, 216.2 (7) [M]+∙.
Supporting Information
| Supporting Information File 1: Spectroscopic data for the synthesis of some alcohols. Quantitative mass efficiency analysis of four alternative alcohol reduction reactions. | ||
| Format: PDF | Size: 326.6 KB | Download |
References
-
Larock, R. C., Ed. Comprehensive organic transformations: a guide to functional group preparations, 2nd ed.; Wiley-VCH: New York, 1999; pp 44–49.
Return to citation in text: [1] -
McCombie, S. W. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, U.K., 1991; Vol. 8, pp 811–833. doi:10.1016/B978-0-08-052349-1.00247-X
Return to citation in text: [1] -
Zard, S. Z. Xanthates and Related Derivatives as Radical Precursors. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1, pp 90–108. doi:10.1002/9783527618293.ch6
Return to citation in text: [1] -
ten Dam, J.; Hanefeld, U. ChemSusChem 2011, 4, 1017–1034. doi:10.1002/cssc.201100162
Return to citation in text: [1] -
Diéguez, H. R.; López, A.; Domingo, V.; Arteaga, J. F.; Dobado, J. A.; Herrador, M. M.; Quílez del Moral, J. F.; Barrero, A. F. J. Am. Chem. Soc. 2010, 132, 254–259. doi:10.1021/ja906083c
Return to citation in text: [1] -
Ledon, H.; Tkatchenko, I.; Young, D. Tetrahedron Lett. 1979, 20, 173–176. doi:10.1016/S0040-4039(01)85916-5
Return to citation in text: [1] -
McMurry, J. E.; Silvestri, M. G.; Fleming, M. P.; Hoz, T.; Grayston, M. W. J. Org. Chem. 1978, 43, 3249–3255. doi:10.1021/jo00411a001
Return to citation in text: [1] -
Sato, F.; Tomuro, Y.; Ishikawa, H.; Oikawa, T.; Sato, M. Chem. Lett. 1980, 9, 103–106. doi:10.1246/cl.1980.103
Return to citation in text: [1] -
Corey, E. J.; Achiwa, K. J. Org. Chem. 1969, 34, 3667–3668. doi:10.1021/jo01263a114
Return to citation in text: [1] -
Crevier, T. J.; Mayer, J. M. J. Am. Chem. Soc. 1997, 119, 8485–8491. doi:10.1021/ja970929s
Return to citation in text: [1] -
Lee, J.-T.; Alper, H. Tetrahedron Lett. 1990, 31, 4101–4104. doi:10.1016/S0040-4039(00)97553-1
Return to citation in text: [1] -
Spiegel, D. A.; Wiberg, K. B.; Schacherer, L. N.; Medeiros, M. R.; Wood, J. L. J. Am. Chem. Soc. 2005, 127, 12513–12515. doi:10.1021/ja052185l
Return to citation in text: [1] -
Zhang, L.; Koreeda, M. J. Am. Chem. Soc. 2004, 126, 13190–13191. doi:10.1021/ja0462777
Return to citation in text: [1] -
Barton, D. H. R.; McCombie, S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574–1585. doi:10.1039/p19750001574
Return to citation in text: [1] -
Barton, D. H. R.; Jang, D. O.; Jaszberenyi, J. C. Synlett 1991, 435–438. doi:10.1055/s-1991-20755
Return to citation in text: [1] -
Barton, D. H. R.; Jang, D. O.; Jaszberenyi, J. C. J. Org. Chem. 1993, 58, 6838–6842. doi:10.1021/jo00076a054
Return to citation in text: [1] -
Barton, D. H. R.; Motherwell, W. B.; Stange, A. Synthesis 1981, 743–745. doi:10.1055/s-1981-29587
Return to citation in text: [1] -
Zard, S. Z. Angew. Chem., Int. Ed. Engl. 1997, 36, 672–685. doi:10.1002/anie.199706721
Return to citation in text: [1] -
Appel, K. E. Drug. Metab. Rev. 2004, 36, 763–786. doi:10.1081/DMR-200033490
Return to citation in text: [1] -
Boyer, I. J. Toxicology 1989, 55, 253–298. doi:10.1016/0300-483X(89)90018-8
Return to citation in text: [1] -
Dopp, E.; Hartmann, L. M.; Florea, A.-M.; Rettenmeier, A. W.; Hirner, A. V. Crit. Rev. Toxicol. 2004, 34, 301–333. doi:10.1080/10408440490270160
Return to citation in text: [1] -
Aloy, J.; Rabaut, C. Bull. Soc. Chim. Fr. 1911, 9, 762–764.
Return to citation in text: [1] -
Aramini, A.; Sablone, M. R.; Bianchini, G.; Amore, A.; Fanì, M.; Perrone, P.; Dolce, A.; Allegretti, M. Tetrahedron 2009, 65, 2015–2021. doi:10.1016/j.tet.2009.01.005
Return to citation in text: [1] -
Dozeman, G. J.; Fiore, P. J.; Puls, T. P.; Walker, J. C. Org. Process Res. Dev. 1997, 1, 137–148. doi:10.1021/op9600419
Return to citation in text: [1] -
Gordon, P. E.; Fry, A. J. Tetrahedron Lett. 2001, 42, 831–833. doi:10.1016/S0040-4039(00)02159-6
Return to citation in text: [1] -
Harvey, R. G.; Leyba, C.; Konieczny, M.; Fu, P. P.; Sukumaran, K. B. J. Org. Chem. 1978, 43, 3423–3425. doi:10.1021/jo00411a048
Return to citation in text: [1] -
Hicks, L. D.; Han, J. K.; Fry, A. J. Tetrahedron Lett. 2000, 41, 7817–7820. doi:10.1016/S0040-4039(00)01359-9
Return to citation in text: [1] -
Marvel, C. S.; Hager, F. D.; Caudle, E. C. Org. Synth. 1923, 3, 45.
Return to citation in text: [1] [2] -
Miescher, K.; Billeter, J. R. Helv. Chim. Acta 1939, 22, 601–610. doi:10.1002/hlca.19390220174
Return to citation in text: [1] -
Platt, K. L.; Oesch, F. J. Org. Chem. 1981, 46, 2601–2603. doi:10.1021/jo00325a041
Return to citation in text: [1] -
Shaw, K. N. F.; Armstrong, M. D.; McMillan, A. J. Org. Chem. 1956, 21, 1149–1151. doi:10.1021/jo01116a023
Return to citation in text: [1] -
Sugita, S.-I.; Toda, S.; Yoshiyasu, T.; Teraji, T. Mol. Cryst. Liq. Cryst. 1993, 237, 399–406. doi:10.1080/10587259308030152
Return to citation in text: [1] -
Czaplicki, S.; Kostanecki, S. T. V.; Lampe, V. Ber. Dtsch. Chem. Ges. 1909, 42, 827–838. doi:10.1002/cber.190904201133
Return to citation in text: [1] -
Milne, J. E.; Storz, T.; Colyer, J. T.; Thiel, O. R.; Dilmeghani Seran, M.; Larsen, R. D.; Murry, J. A. J. Org. Chem. 2011, 76, 9519–9524. doi:10.1021/jo2018087
Return to citation in text: [1] -
Wu, G. G.; Chen, F. X.; LaFrance, D.; Liu, Z.; Greene, S. G.; Wong, Y.-S.; Xie, J. Org. Lett. 2011, 13, 5220–5223. doi:10.1021/ol102174w
Return to citation in text: [1] -
Deno, N. C.; Friedman, N.; Hodge, J. D.; MacKay, F. P.; Saines, G. J. Am. Chem. Soc. 1962, 84, 4713–4715. doi:10.1021/ja00883a019
Return to citation in text: [1] -
Gordon, P. E.; Fry, A. J.; Hicks, L. D. ARKIVOC 2005, vi, 393–400.
Return to citation in text: [1] -
Ogg, R. A., Jr. J. Am. Chem. Soc. 1934, 56, 526–536. doi:10.1021/ja01318a007
Return to citation in text: [1] -
Eissen, M.; Metzger, J. O. Chem.–Eur. J. 2002, 8, 3580–3585. doi:10.1002/1521-3765(20020816)8:16<3580::AID-CHEM3580>3.0.CO;2-J
Return to citation in text: [1] -
Takahashi, M.; McLaughlin, M.; Micalizio, G. C. Angew. Chem., Int. Ed. 2009, 48, 3648–3652. doi:10.1002/anie.200900236
Return to citation in text: [1] -
Zhou, C.; Wang, Z. Synthesis 2005, 1649–1655. doi:10.1055/s-2005-865293
Return to citation in text: [1]
| 1. | Larock, R. C., Ed. Comprehensive organic transformations: a guide to functional group preparations, 2nd ed.; Wiley-VCH: New York, 1999; pp 44–49. |
| 2. | McCombie, S. W. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, U.K., 1991; Vol. 8, pp 811–833. doi:10.1016/B978-0-08-052349-1.00247-X |
| 3. | Zard, S. Z. Xanthates and Related Derivatives as Radical Precursors. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1, pp 90–108. doi:10.1002/9783527618293.ch6 |
| 4. | ten Dam, J.; Hanefeld, U. ChemSusChem 2011, 4, 1017–1034. doi:10.1002/cssc.201100162 |
| 15. | Barton, D. H. R.; Jang, D. O.; Jaszberenyi, J. C. Synlett 1991, 435–438. doi:10.1055/s-1991-20755 |
| 16. | Barton, D. H. R.; Jang, D. O.; Jaszberenyi, J. C. J. Org. Chem. 1993, 58, 6838–6842. doi:10.1021/jo00076a054 |
| 17. | Barton, D. H. R.; Motherwell, W. B.; Stange, A. Synthesis 1981, 743–745. doi:10.1055/s-1981-29587 |
| 18. | Zard, S. Z. Angew. Chem., Int. Ed. Engl. 1997, 36, 672–685. doi:10.1002/anie.199706721 |
| 14. | Barton, D. H. R.; McCombie, S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574–1585. doi:10.1039/p19750001574 |
| 9. | Corey, E. J.; Achiwa, K. J. Org. Chem. 1969, 34, 3667–3668. doi:10.1021/jo01263a114 |
| 10. | Crevier, T. J.; Mayer, J. M. J. Am. Chem. Soc. 1997, 119, 8485–8491. doi:10.1021/ja970929s |
| 11. | Lee, J.-T.; Alper, H. Tetrahedron Lett. 1990, 31, 4101–4104. doi:10.1016/S0040-4039(00)97553-1 |
| 12. | Spiegel, D. A.; Wiberg, K. B.; Schacherer, L. N.; Medeiros, M. R.; Wood, J. L. J. Am. Chem. Soc. 2005, 127, 12513–12515. doi:10.1021/ja052185l |
| 13. | Zhang, L.; Koreeda, M. J. Am. Chem. Soc. 2004, 126, 13190–13191. doi:10.1021/ja0462777 |
| 5. | Diéguez, H. R.; López, A.; Domingo, V.; Arteaga, J. F.; Dobado, J. A.; Herrador, M. M.; Quílez del Moral, J. F.; Barrero, A. F. J. Am. Chem. Soc. 2010, 132, 254–259. doi:10.1021/ja906083c |
| 6. | Ledon, H.; Tkatchenko, I.; Young, D. Tetrahedron Lett. 1979, 20, 173–176. doi:10.1016/S0040-4039(01)85916-5 |
| 7. | McMurry, J. E.; Silvestri, M. G.; Fleming, M. P.; Hoz, T.; Grayston, M. W. J. Org. Chem. 1978, 43, 3249–3255. doi:10.1021/jo00411a001 |
| 8. | Sato, F.; Tomuro, Y.; Ishikawa, H.; Oikawa, T.; Sato, M. Chem. Lett. 1980, 9, 103–106. doi:10.1246/cl.1980.103 |
| 36. | Deno, N. C.; Friedman, N.; Hodge, J. D.; MacKay, F. P.; Saines, G. J. Am. Chem. Soc. 1962, 84, 4713–4715. doi:10.1021/ja00883a019 |
| 37. | Gordon, P. E.; Fry, A. J.; Hicks, L. D. ARKIVOC 2005, vi, 393–400. |
| 38. | Ogg, R. A., Jr. J. Am. Chem. Soc. 1934, 56, 526–536. doi:10.1021/ja01318a007 |
| 39. | Eissen, M.; Metzger, J. O. Chem.–Eur. J. 2002, 8, 3580–3585. doi:10.1002/1521-3765(20020816)8:16<3580::AID-CHEM3580>3.0.CO;2-J |
| 33. | Czaplicki, S.; Kostanecki, S. T. V.; Lampe, V. Ber. Dtsch. Chem. Ges. 1909, 42, 827–838. doi:10.1002/cber.190904201133 |
| 34. | Milne, J. E.; Storz, T.; Colyer, J. T.; Thiel, O. R.; Dilmeghani Seran, M.; Larsen, R. D.; Murry, J. A. J. Org. Chem. 2011, 76, 9519–9524. doi:10.1021/jo2018087 |
| 35. | Wu, G. G.; Chen, F. X.; LaFrance, D.; Liu, Z.; Greene, S. G.; Wong, Y.-S.; Xie, J. Org. Lett. 2011, 13, 5220–5223. doi:10.1021/ol102174w |
| 40. | Takahashi, M.; McLaughlin, M.; Micalizio, G. C. Angew. Chem., Int. Ed. 2009, 48, 3648–3652. doi:10.1002/anie.200900236 |
| 22. | Aloy, J.; Rabaut, C. Bull. Soc. Chim. Fr. 1911, 9, 762–764. |
| 23. | Aramini, A.; Sablone, M. R.; Bianchini, G.; Amore, A.; Fanì, M.; Perrone, P.; Dolce, A.; Allegretti, M. Tetrahedron 2009, 65, 2015–2021. doi:10.1016/j.tet.2009.01.005 |
| 24. | Dozeman, G. J.; Fiore, P. J.; Puls, T. P.; Walker, J. C. Org. Process Res. Dev. 1997, 1, 137–148. doi:10.1021/op9600419 |
| 25. | Gordon, P. E.; Fry, A. J. Tetrahedron Lett. 2001, 42, 831–833. doi:10.1016/S0040-4039(00)02159-6 |
| 26. | Harvey, R. G.; Leyba, C.; Konieczny, M.; Fu, P. P.; Sukumaran, K. B. J. Org. Chem. 1978, 43, 3423–3425. doi:10.1021/jo00411a048 |
| 27. | Hicks, L. D.; Han, J. K.; Fry, A. J. Tetrahedron Lett. 2000, 41, 7817–7820. doi:10.1016/S0040-4039(00)01359-9 |
| 28. | Marvel, C. S.; Hager, F. D.; Caudle, E. C. Org. Synth. 1923, 3, 45. |
| 29. | Miescher, K.; Billeter, J. R. Helv. Chim. Acta 1939, 22, 601–610. doi:10.1002/hlca.19390220174 |
| 30. | Platt, K. L.; Oesch, F. J. Org. Chem. 1981, 46, 2601–2603. doi:10.1021/jo00325a041 |
| 31. | Shaw, K. N. F.; Armstrong, M. D.; McMillan, A. J. Org. Chem. 1956, 21, 1149–1151. doi:10.1021/jo01116a023 |
| 32. | Sugita, S.-I.; Toda, S.; Yoshiyasu, T.; Teraji, T. Mol. Cryst. Liq. Cryst. 1993, 237, 399–406. doi:10.1080/10587259308030152 |
| 19. | Appel, K. E. Drug. Metab. Rev. 2004, 36, 763–786. doi:10.1081/DMR-200033490 |
| 20. | Boyer, I. J. Toxicology 1989, 55, 253–298. doi:10.1016/0300-483X(89)90018-8 |
| 21. | Dopp, E.; Hartmann, L. M.; Florea, A.-M.; Rettenmeier, A. W.; Hirner, A. V. Crit. Rev. Toxicol. 2004, 34, 301–333. doi:10.1080/10408440490270160 |
© 2012 Dobmeier et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)