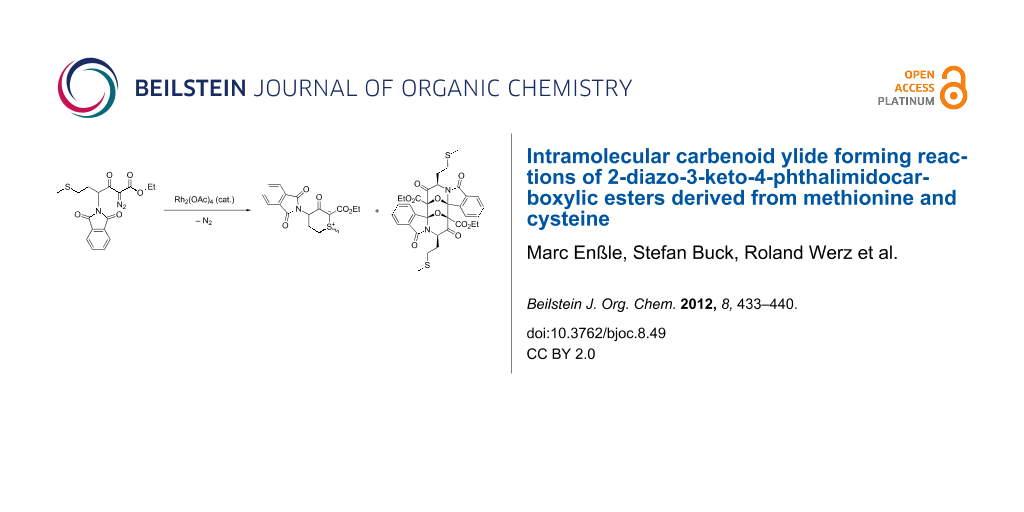
Abstract
Methionine, S-benzylcysteine and S-allylcysteine were converted into 2-diazo-3-oxo-4-phthalimidocarboxylic esters 8a–c in three steps. Upon rhodium-catalysed dediazoniation, two intramolecular carbenoid reactions competed, namely the formation of a cyclic sulfonium ylide and that of a six-ring carbonyl ylide. The S-methyl and S-benzyl ylides 12a and b could be isolated, while S-allyl ylide 12c underwent a [2,3]-sigmatropic rearrangement. The short-lived carbonyl ylides derived from methionine and S-benzylcysteine formed head-to-tail dimers by a [3 + 3]-cycloaddition and could be trapped with external dipolarophiles, while the S-allyl derivative 14c yielded the pentacyclic compound 17 by an intramolecular [3 + 2]-cycloaddition reaction.

Graphical Abstract
Introduction
The synthetic potential of diazo compounds, in particular of α-diazoketones and α-diazoesters, is greatly widened by the ability of the derived carbene or metal-carbene intermediates to undergo inter- and intramolecular formation of N-, O-, S- and other ylides [1,2]. The benefit of these transformations, which are usually performed with rhodium- or copper-based catalysts, is given by subsequent rearrangement or addition reactions of the reactive ylides; several recent reviews [3-8] illustrate the wide range of applications. As far as sulfonium ylides are concerned, thermally induced isomerisation, that is the 1,2-shift of a substituent (Stevens rearrangement) and [2,3]-sigmatropic rearrangement of allylsulfonium ylides [9-11], and the use as C1 building blocks in epoxidation, aziridination and olefination reactions [12,13] are common reaction channels. The intramolecular formation of sulfonium ylides from α-diazocarbonyl compounds tethered with alkylthio or arylthio groups has been studied by the research groups of Davies [14], Moody [15], and West [16]. From α-diazo-β-ketoesters 1, stable four- to seven-membered cyclic ylides 2 were obtained (Scheme 1); in the case of R2 = allyl, however, the ylides underwent a spontaneous [2,3]-sigmatropic rearrangement.
![[1860-5397-8-49-i1]](/bjoc/content/inline/1860-5397-8-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of cyclic sulfonium ylides 2; n = 0–3.
Scheme 1: Synthesis of cyclic sulfonium ylides 2; n = 0–3.
On the other hand, the conversion of methionine-derived diazoketone 3 into the cyclic sulfonium ylide 4 was not accomplished through the carbenoid route, but rather by cyclisation of a diazonium ion followed by deprotonation of the resulting sulfonium ion (Scheme 2) [17].
![[1860-5397-8-49-i2]](/bjoc/content/inline/1860-5397-8-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Non-carbenoid formation of sulfonium ylide 4.
Scheme 2: Non-carbenoid formation of sulfonium ylide 4.
A diazoketone such as 3 has the structural prerequisites to undergo two types of intramolecular carbenoid ylide-forming reactions, yielding either a cyclic sulfonium ylide or a six-ring carbonyl ylide. In fact, it has been found that Rh(II)-catalyzed dediazoniation of α-diazo-β-ketoesters with γ-phthalimido [18,19] or related [20] substituents gives rise to cyclic carbonyl ylides, which were trapped by intermolecular cycloaddition reactions. In this paper, we report that the formation of sulfonium as well as carbonyl ylides are indeed competing pathways in the Rh(II)-catalyzed dediazoniation of phthaloyl-protected γ-amino-α-diazo-β-ketoesters derived from methionine, S-benzylcysteine, and S-allylcysteine.
Results and Discussion
Synthesis of diazoesters
The 2-diazo-3-oxo-4-phthalimidocarboxylic esters 8a–c were prepared from L-methionine (5a), S-benzyl-L-cysteine (5b) and S-allyl-L-cysteine (5c), respectively, in a three-step sequence (Scheme 3) applied by us previously for other α-aminoacids [18]. Acids 5a–c were converted into their phthalimido-substituted derivatives 6 followed by conversion into β-ketoesters 7 (yields: 30% (7a), 60% (7b) and 19% (7c)) and diazo group transfer to give diazoesters 8 (61–99%). Phthalimido-free diazoesters 11a and b, which were desired for comparative reactivity studies, were prepared analogously; it should be noted that different approaches were taken to obtain the methyl ester analogue of 11b and other similar sulfur-containing α-diazo-β-ketoesters [14-16]. We found that diazoester 8a can also be prepared by acylation of ethyl diazoacetate (two equivalents, one equivalent serving to trap HCl) with the acid chloride of 6a. Although the yield was modest (28%), it was still better and gave fewer byproducts than the β-ketoester route.
![[1860-5397-8-49-i3]](/bjoc/content/inline/1860-5397-8-49-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Conditions: (a) phthalic anhydride, NEt3 (10 mol %), toluene, reflux, 2 h; (b) 1. carbonyldiimidazole, THF, 16 h; 2. MgCl2, NEt3, HOOC–CH2CO2Et, 3.5 h; (c) p-tosyl azide, NEt3, acetonitrile, rt, 16 h (with 7a) or imidazole-1-sulfonyl azide·HCl, NEt3, CH2Cl2, reflux, 16 h (with 7b,c and 10). Alternative synthesis of 8a: 1. 6a, SOCl2 (10 equiv), CH2Cl2, reflux, 2 h; 2. N2=CHCO2Et (2 equiv), rt, 4 days.
Scheme 3: Conditions: (a) phthalic anhydride, NEt3 (10 mol %), toluene, reflux, 2 h; (b) 1. carbonyldiimidazo...
The phthaloylation of 5a–c was achieved with phthalic anhydride in the presence of a catalytic amount of triethylamine to lower the reaction temperature, so that the reaction could be performed in toluene under reflux. For phenylalanine, it was reported that no racemisation occurred under these conditions [21]. However, with some other α-aminoacids we have noted a small degree of racemisation [18]. In this work, we can confirm the reported specific rotation value for N,N-phthaloyl-L-methionine (6a), but racemisation occurred to a significant extent in the case of S-benzyl-N,N-phthaloyl-L-cysteine (6b) (Supporting Information File 1). Practically complete racemisation took place during conversion of N-protected aminoacids 6a–c to the β-ketoesters 7a–c, and therefore, diazoesters 8a–c were also obtained in (almost) racemic form. As a consequence, diazoester 8a was prepared subsequently from DL-methionine. On the other hand, the preparation of N-phthaloylated diazoester 8a via the aminoacid chloride route (Scheme 3) seems to occur with only a little racemisation. A sample of 8a, which was prepared from a slightly racemised sample of 6a (HPLC: 90:10 enantiomeric ratio of the derived methyl ester, which was prepared from 6a and dry methanol in the presence of excess chlorotrimethylsilane) on the latter route, had an enantiomeric ratio of 83:17.
Rhodium-catalysed ylide-forming reactions
A sample of racemic diazoester 8a, prepared from DL-methionine, was exposed to a catalytic amount of dirhodium tetraacetate in boiling benzene. Two products were isolated and identified, the six-membered cyclic sulfonium ylide 12a (13% yield) and the carbonyl ylide dimer 13a (43%), (Scheme 4). In an analogous manner, the five-membered cyclic sulfonium ylide 12b and polyheterocycle 13b were formed from diazoester 8b in yields of 37 and 25%, respectively (yields determined by 1H NMR integration on the crude product mixture). In contrast to these successful transformations, we found that α-diazoketone 3 (Scheme 2) was smoothly decomposed by a catalytic amount of Rh2(OAc)4 or Cu(I) triflate (e.g., 1 mol % of Rh2(OAc)4, CH2Cl2, 40 °C, 17 h), but a complex mixture of unidentified products was formed.
![[1860-5397-8-49-i4]](/bjoc/content/inline/1860-5397-8-49-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Rh(II)-catalysed carbenoid reactions of diazoesters 8a,b.
Scheme 4: Rh(II)-catalysed carbenoid reactions of diazoesters 8a,b.
The cyclic sulfonium ylides 12a,b were both obtained as a mixture of two diastereomers. For the six-ring ylide 12a (isomer ratio A:B = 4:1) a stereochemical assignment was made based on 1H NMR studies (Figure 1 and Table 1). We propose that the major isomer 12aA assumes a cyclohexene-like half-chair conformation, and the minor isomer 12aB, which has the opposite configuration at the phthalimido-substituted ring position, exists in a twisted-boat form. In both cases, the S-methyl group occupies the (pseudo-)axial position and the sterically demanding phthalimido group is in the (pseudo-)equatorial position. These conformations fit the observed nuclear Overhauser effects as well as the 3J coupling constants. Crystal structure determinations of an acyclic [22] and a five-membered cyclic [23] sulfonium ylide containing the R2S+–C−(COOR or COCH3)–C(=O)R moiety have shown that the sulfur atom is pyramidalised and the negative charge is delocalised over the π-system of the planar oxoenolate unit. This geometry agrees with the proposed conformation of 12aA. In the twisted-boat structure of 12B, the negative charge may be stabilised mainly by the ester group and less so by the adjacent keto group. The axial position of the methyl group at the pyramidalised sulfur atom is remarkable. In this geometry, however, p(Cylide)–σ*(S–C) conjugation of the unshared pair of electrons at the ylidic carbon atom would be feasible. While this interaction is a key stabilisation factor for the parent sulfonium ylides (e.g., H2S–CH2 and Me2S–CH2) according to theoretical calculations [24], its energetic contribution in the case of sulfonium ylides stabilised by acceptor substituents is likely to be small.
![[1860-5397-8-49-1]](/bjoc/content/figures/1860-5397-8-49-1.svg?scale=1.6&max-width=1024&background=FFFFFF)
Figure 1: Proposed relative configurations of the diastereomeric cyclic sulfonium ylides 12aA and 12aB. 1H NMR shifts δH (ppm) and NOE relationships are shown (E = CO2Et).
Figure 1: Proposed relative configurations of the diastereomeric cyclic sulfonium ylides 12aA and 12aB. 1H NM...
Table 1: 1H NMR data for isomers A (major) and B (minor) of cyclic sulfonium ylide 12a (DMSO-d6, 400.13 MHz).
| Assignment |
Isomer A
δ (ppm), multiplicity, J (Hz)a |
Isomer B
δ (ppm), multiplicity, J (Hz)a |
|---|---|---|
| OCH2CH3 | 1.13, t, J = 7.1 | 1.14, t, J = 7.1 |
| SCH3 | 2.80, s | 2.83, s |
| SCHeq b | 3.41–3.46, m, 2J = 14.1, 3J = 2.8, 2.8 | 3.41–3.46, ddd, 2J = 13.3, 3J = 9.7, 4.1 |
| SCHax b | 3.77–3.83, m, 2J ≈ 3J ≈ 14.1, 3J = 2.7 | 3.77–3.83, ddd, 2J = 13.3, 3J = 7.3, 4.3 |
| SCH2CHeq | 2.35–2.41 dddd, 2J = 14.1, 3J = 5.7, 2.8, 2.8 | 2.57–2.72, m |
| SCH2CHax | 3.05, dddd, 2J ≈ 3J ≈ 14.1, 3J = 12.7, 3J = 2.9 | 2.57–2.72, m |
| NCHCO | 4.65, dd, 3J = 12.7, 5.7 | 4.75, dd, 3J = 9.1, 5.6 |
| OCH2 | 3.94–4.07, m (both isomers) | |
| CHaryl | 7.80–7.96, broadened “s” (both isomers) | |
aCoupling constants are given without sign.
bThe multiplet signals of SCHeq and SCHax of the two isomers coincided completely. A magnetisation transfer experiment (TOCSY) allowed visualisation of the individual signal patterns.
The oxazapolycycles 13a and b are head-to-tail dimers of carbonyl ylides, resulting from a [3 + 3]-cycloaddition. Their constitution and relative stereochemistry was assigned by NMR comparison with the corresponding alanine-derived ring system, for which a structural proof was furnished by X-ray crystal-structure determination [18]. Due to the symmetry of the dimers, the NMR spectra display signals for only half of the total number of carbon and hydrogen nuclei. Characteristics are the signals for the NCH proton (13a: δ = 4.60 ppm; 13b: δ = 4.67 ppm) and the two bridgehead carbons of the epoxy bridge (13a: δ = 86.60 and 94.13 ppm; 13b: δ = 86.75 and 94.04 ppm). While a CI mass spectrum of 13b showed a weak molecular-ion peak for the dimer (the base peak representing the monomer unit), in the case of 13a, even under these mild conditions of chemical ionisation, the spectrum showed no peak at m/z greater than that for the monomer, which was again the base peak. The relative stereochemistry shown in Scheme 4, with the two epoxy bridges in syn relationship and the mercaptoalkyl substituent in the (equatorial) exo position, is explained by an endo transition state 15 for the [3 + 3]-cycloaddition, in which two cyclic carbonyl ylide molecules 14 approach each other with their sterically less-occupied faces (Scheme 5). Our findings on carbonyl ylide dimers reported here and in a preceding paper [18] tie in with earlier reports on the [3 + 3]-cyclodimerisation of carbonyl ylides of the 2-benzopyrylium-4-olate type [25,26].
![[1860-5397-8-49-i5]](/bjoc/content/inline/1860-5397-8-49-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Endo transition state for [3 + 3]-dimerisation of carbonyl ylide 14.
Scheme 5: Endo transition state for [3 + 3]-dimerisation of carbonyl ylide 14.
A different product pattern results from the rhodium(II)-catalysed dediazoniation of S-allyl-substituted diazoester 8c (Scheme 6). In this case, the simultaneous formation of five-ring sulfonium ylide 12c and six-ring carbonyl ylide 14c is immediately followed by intramolecular pericyclic reactions with participation of the allylic π-system. Sulfonium ylide 12c rearranges by the expected [2,3]-sigmatropic rearrangement [3,10,16] to form tetrahydrothiophene 16 (31% yield, two diastereomers), and carbonyl ylide 14c is trapped by an intramolecular [3 + 2]-cycloaddition to give the pentacyclic compound 17 (35% yield). The regioselectivity of the latter reaction was established by NMR studies: after assignment of proton resonances from HMBC spectra, a NOESY NMR experiment indicated the correlation shown in Scheme 6. Intramolecular [3 + 2]-cycloaddition reactions of a carbonyl ylide with a nonactivated but suitably positioned olefinic bond have been known for some time [3,27-35] and can be used for the construction of multicyclic molecular frameworks (see also those carbonyl ylides that are part of an isomünchnone ring system [36-40]). In almost all cases encountered so far, the olefinic dipolarophile was tethered directly to a terminus of the carbonyl ylide dipole, and therefore the cycloaddition step gave rise to an annelated bicyclic substructure. In contrast, in our case as well as in [29], the olefinic dipolarophile is found as a substituent at a remote ring position of the cyclic carbonyl ylide, such that the intramolecular 1,3-dipolar cycloaddition generates a bridged oxabicyclo[3.2.1]octane substructure.
![[1860-5397-8-49-i6]](/bjoc/content/inline/1860-5397-8-49-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Rh(II)-catalysed carbenoid reactions of diazoester 8c.
Scheme 6: Rh(II)-catalysed carbenoid reactions of diazoester 8c.
Trapping of carbonyl ylides with dipolarophiles
In situ generated carbonyl ylides can be intercepted not only in intramolecular (as discussed above) but also in intermolecular [3 + 2] cycloaddition reactions (see, for example, [3,18-20,41,42]). By analogy with other 2-diazo-3-oxo-4-phthalimidocarboxylic esters [18,19], treatment of diazoester 8a with catalytic Rh2(OAc)4 in the presence of an equimolar amount of N-phenylmaleimide (NPI) or dimethyl acetylenedicarboxylate (DMAD) produced the cycloaddition products 18 and 19, respectively (Scheme 7). The signal sets of sulfonium ylides 12aA and 12aB were absent from the 1H NMR spectra of the crude product mixture. For adduct 18, only one diastereomer was obtained (38% yield after workup), which, according to the observed NOE effects, has the pyrrolidinedione ring and the (CH2)2SCH3 chain in exo positions. This means that the cycloaddition has gone through an exo transition state with the dipolarophile approaching the carbonyl ylide from the face opposite to the sulfur-containing alkyl chain. In contrast, the NPI adducts of analogous carbonyl ylides derived from norleucine and alanine (14, R = n-C4H9 and CH3, respectively), were obtained as two diastereomers (the NCHR epimers) in approximately equal ratio [18]. The DMAD adduct 19 was also formed as a 2:1 mixture of two diastereomers; unfortunately, the highly viscous oil obtained could not be purified completely.
![[1860-5397-8-49-i7]](/bjoc/content/inline/1860-5397-8-49-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Tandem cyclisation/intermolecular cycloaddition of diazoester 8a. Conditions: (a) Rh2(OAc)4 (3 mol %), N-phenylmaleimide or DMAD (1 equiv), benzene, 80 °C, 4 h.
Scheme 7: Tandem cyclisation/intermolecular cycloaddition of diazoester 8a. Conditions: (a) Rh2(OAc)4 (3 mol ...
Rhodium-catalysed carbenoid reactions of other mercapto-functionalised diazoesters
With respect to sulfonium ylide formation, aminoacid-derived diazoesters 8a–c behave like the analogous mercapto-functionalised diazoesters that are devoid of the phthalimido group (Scheme 8). Thus, rhodium(II)-catalysed decomposition of 2-diazo-6-methylthio-3-oxohexanoate 11a gave the stable six-ring sulfonium ylide 20 in practically quantitative yield. It has recently been reported that ruthenium(II) porphyrins are suitable catalysts for carbenoid sulfonium ylide formation as well [43]. In continuation of our comparative studies of dirhodium(II,II) tetracarboxylate and tetracarbonyldiruthenium(I,I) complexes [44], we found that the saccharinato complex [Ru2(CO)5[μ-sac)2]2 [45], the acetato complex [Ru2(CO)4(μ-OAc)2]n and the trinuclear ruthenium(0) complex Ru3(CO)12 (3 mol % of catalyst in each case) gave yields of 74, 69, and 49% for the conversion of 11a to 20.
![[1860-5397-8-49-i8]](/bjoc/content/inline/1860-5397-8-49-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Carbenoid formation of sulfonium ylides from diazoesters 11a,b. Conditions: (a) Rh2(OAc)4 (3 mol %), benzene, 80 °C, 2 h (11a) or 30 min (11b).
Scheme 8: Carbenoid formation of sulfonium ylides from diazoesters 11a,b. Conditions: (a) Rh2(OAc)4 (3 mol %)...
The rhodium-catalysed formation of a stable five-ring sulfonium ylide from the methyl ester analogue of diazoester 11b has previously been reported [15]. We confirm these results with 11b; however, in addition to sulfonium ylide 21 we also isolated the dienol 22 as a minor byproduct. The latter compound likely results from a thermally induced ring-opening β-elimination reaction of ylide 21, as was previously reported for other cyclic [14] and acyclic [46] sulfonium ylides.
Conclusion
Metal-carbene intermediates derived from α-diazoesters and α-diazo-β-ketoesters are known for their high reactivity towards a range of functional groups. It is therefore interesting to gather information about the chemoselectivity of these reactive intermediates. For intramolecular reactions, the ring size of the products can make an additional contribution to the observed chemoselectivity. In this study, we have addressed the competition between two intramolecular ylide-forming pathways, namely the formation of cyclic sulfonium and carbonyl ylides. To this end, 2-diazo-3-oxo-4-phthalimidocarboxylic esters 8a–c were prepared from the mercapto-functionalised aminoacids methionine, S-benzylcysteine, and S-allylcysteine. Rhodium(II)-catalysed decomposition of the diazoesters was found to produce both six- or five-membered cyclic sulfonium ylides and six-membered carbonyl ylides. In a qualitative manner, it can be stated that in the case of 8a formation of the six-membered cyclic carbonyl ylide 14a clearly supersedes the formation of the six-membered sulfonium ylide 12a, although the structurally analogous cyclic sulfonium ylide 20 is formed quantitatively from diazoester 11a, which bears no phthalimido group. This result is confirmed by the absence of 12a from the reactions in which the carbonyl ylide was intercepted by intermolecular [3 + 2] cycloaddition with the dipolarophiles NPI and DMAD (notice that carbonyl ylide dimer 13a also results from an intermolecular cycloaddition reaction). With the cysteine-derived diazoesters 8b and c, on the other hand, it appears that the formation of the five-membered cyclic sulfonium ylides 12b and c and of the six-membered cyclic carbonyl ylides 14b and c is about equally efficient. Interestingly, the S-allyl group spontaneously transforms both types of ylides into non-ylidic products, namely by [2,3]-sigmatropic rearrangement for the S-ylide and intramolecular [3 + 2]-cycloaddition for the carbonyl ylide.
Supporting Information
The supporting information contains experimental procedures and characterisation details for the synthesised compounds.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 285.9 KB | Download |
References
-
Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; John Wiley & Sons: New York, 1998; Chapter 7.
Return to citation in text: [1] -
Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263–309. doi:10.1021/cr00003a001
Return to citation in text: [1] -
Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223–270. doi:10.1021/cr950022h
Return to citation in text: [1] [2] [3] [4] -
Doyle, M. P.; Forbes, D. C. Chem. Rev. 1998, 98, 911–936. doi:10.1021/cr940066a
Return to citation in text: [1] -
Davies, H. M. L.; Beckwith, R. E. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217
Return to citation in text: [1] -
Wee, A. G. H. Curr. Org. Synth. 2006, 3, 499–555. doi:10.2174/157017906778699512
Return to citation in text: [1] -
Ferreira, V. F. Curr. Org. Chem. 2007, 11, 177–193. doi:10.2174/138527207779316462
Return to citation in text: [1] -
Zhang, Z.; Wang, J. Tetrahedron 2008, 64, 6577–6605. doi:10.1016/j.tet.2008.04.074
Return to citation in text: [1] -
Lakeev, S. N.; Maydanova, I. O.; Galin, F. Z.; Tolstikov, G. A. Russ. Chem. Rev. 2001, 70, 655–672. doi:10.1070/RC2001v070n08ABEH000645
Return to citation in text: [1] -
Sweeney, J. G. Chem. Soc. Rev. 2009, 38, 1027–1038. doi:10.1039/b604828p
Return to citation in text: [1] [2] -
Zhang, Y.; Wang, J. Coord. Chem. Rev. 2010, 254, 941–953. doi:10.1016/j.ccr.2009.12.005
Return to citation in text: [1] -
Li, A.-H.; Dai, L.-X.; Aggarwal, V. K. Chem. Rev. 1997, 97, 2341–2372. doi:10.1021/cr960411r
Return to citation in text: [1] -
Aggarwal, V. K.; Winn, C. L. Acc. Chem. Res. 2004, 37, 611–620. doi:10.1021/ar030045f
Return to citation in text: [1] -
Davies, H. M. L.; Crisco, L. Van T. Tetrahedron Lett. 1987, 28, 371–374. doi:10.1016/S0040-4039(00)95731-9
Return to citation in text: [1] [2] [3] -
Moody, C. J.; Taylor, R. J. Tetrahedron 1990, 6501–6524. doi:10.1016/S0040-4020(01)96017-X
Return to citation in text: [1] [2] [3] -
Murphy, G. K.; West, F. G. Org. Lett. 2006, 8, 4359–4361. doi:10.1021/ol061772o
Return to citation in text: [1] [2] [3] -
Tolstikov, G. A.; Galin, F. Z.; Lakeev, S. N. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1989, 32, 880.
Return to citation in text: [1] -
Enßle, M.; Buck, S.; Werz, R.; Maas, G. ARKIVOC 2012, (iii), 149–171.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Padwa, A.; Dean, D. C.; Hertzog, D. L.; Nadler, W. R.; Zhi, L. Tetrahedron 1992, 48, 7565–7580. doi:10.1016/S0040-4020(01)90369-2
Return to citation in text: [1] [2] [3] -
Nikolaev, V. V.; Krylov, I. S.; Schulze, B.; Rodina, L. L. Russ. J. Org. Chem. 2005, 41, 784–786. doi:10.1007/s11178-005-0246-3
Return to citation in text: [1] [2] -
Bose, A. K.; Greer, F.; Price, C. C. J. Org. Chem. 1958, 23, 1335–1338. doi:10.1021/jo01103a025
Return to citation in text: [1] -
Yufit, D. S.; Struchkov, Y. T.; Kozhushkov, S. I.; Zefirov, P. S. Russ. Chem. Bull. 1991, 40, 68–73. doi:10.1007/BF00959632
Return to citation in text: [1] -
Igarashi, J.-e.; Watanabe, N.; Konoshita, T.; Ogata, K.; Furukawa, S. Chem. Pharm. Bull. 1990, 38, 329–335. doi:10.1248/cpb.38.329
Return to citation in text: [1] -
Standard, J. M.; Copack, B. A.; Johnson, T. K.; Przybyla, D. E.; Graham, S. R.; Steidl, R. J. J. Phys. Chem. A 2008, 112, 336–341. doi:10.1021/jp077013k
Return to citation in text: [1] -
Zimmerman, H. E.; Simkin, R. D. Tetrahedron Lett. 1964, 5, 1847–1851. doi:10.1016/S0040-4039(00)75116-1
Return to citation in text: [1] -
Nilsen, B. J.; Undheim, K. Acta Chem. Scand. 1976, 30, 619–623. doi:10.3891/acta.chem.scand.30b-0619
Return to citation in text: [1] -
Weingarten, M. D.; Prein, M.; Price, A. T.; Snyder, J. P.; Padwa, A. J. Org. Chem. 1997, 62, 2001–2010. doi:10.1021/jo962184z
Return to citation in text: [1] -
Hodgson, D. M.; Stupple, P. A.; Pierard, F. Y. T. M.; Labande, A. H.; Johnstone, C. Chem.–Eur. J. 2001, 7, 4465–4476. doi:10.1002/1521-3765(20011015)7:20<4465::AID-CHEM4465>3.0.CO;2-W
Return to citation in text: [1] -
Kim, C. H.; Jang, K. P.; Choi, S. Y.; Chung, Y. K.; Lee, E. Angew. Chem., Int. Ed. 2008, 120, 4073–4075. doi:10.1002/anie.200800568
Return to citation in text: [1] [2] -
Mejia-Oneto, J. M.; Padwa, A. Tetrahedron Lett. 2004, 45, 9115–9118. doi:10.1016/j.tetlet.2004.10.018
Return to citation in text: [1] -
Hodgson, D. M.; Angrish, D.; Labande, A. H. Chem. Commun. 2006, 627–628. doi:10.1039/b515943a
Return to citation in text: [1] -
Padwa, A.; Chughtai, M. J.; Boonsombat, J.; Rashatasakhon, P. Tetrahedron 2008, 64, 4758–4767. doi:10.1016/j.tet.2008.01.129
Return to citation in text: [1] -
Oh, C. H.; Lee, J. H.; Lee, S. J.; Kim, J. I.; Hong, C. S. Angew. Chem., Int. Ed. 2008, 120, 7615–7617. doi:10.1002/anie.200802425
Return to citation in text: [1] -
Hodgson, D. M.; Glen, R.; Redgrave, A. J. Tetrahedron: Asymmetry 2009, 20, 754–757. doi:10.1016/j.tetasy.2009.02.031
Return to citation in text: [1] -
Padwa, A. Tetrahedron 2011, 67, 8057–8072. doi:10.1016/j.tet.2011.07.009
Return to citation in text: [1] -
Padwa, A.; Hertzog, D. L.; Nadler, W. R.; Osterhout, M. H.; Price, A. T. J. Org. Chem. 1994, 59, 1418–1427. doi:10.1021/jo00085a035
Return to citation in text: [1] -
Padwa, A.; Heidelbaugh, T. M.; Kuethe, J. T. J. Org. Chem. 1999, 64, 2038–2049. doi:10.1021/jo982315r
Return to citation in text: [1] -
Padwa, A.; Bur, S. K. Tetrahedron 2007, 63, 5341–5378. doi:10.1016/j.tet.2007.03.158
Return to citation in text: [1] -
England, D. B.; Eagan, J. M.; Merey, G.; Anac, O.; Padwa, A. Tetrahedron 2008, 64, 988–1001. doi:10.1016/j.tet.2007.10.038
Return to citation in text: [1] -
Li, H.; Cheng, B.; Boonnak, N.; Padwa, A. Tetrahedron 2011, 67, 9829–9836. doi:10.1016/j.tet.2011.09.118
Return to citation in text: [1] -
Mehta, G.; Muthusamy, S. Tetrahedron 2002, 58, 9477–9504. doi:10.1016/S0040-4020(02)01187-0
Return to citation in text: [1] -
Zhou, C.-Y.; Chan, P. W. H.; Yu, W.-Y.; Che, C.-M. Synthesis 2003, 1403–1412. doi:10.1055/s-2003-40187
Return to citation in text: [1] -
Zhou, C.-Y.; Huang, J.-S.; Che, C.-M. Synlett 2010, 2681–2700. doi:10.1055/s-0030-1259017
Return to citation in text: [1] -
Grohmann, M.; Buck, S.; Schäffler, L.; Maas, G. Adv. Synth. Catal. 2006, 348, 2203–2211. doi:10.1002/adsc.200606108
Return to citation in text: [1] -
Buck, S.; Maas, G. J. Organomet. Chem. 2006, 691, 2774–2784. doi:10.1016/j.jorganchem.2006.02.013
Return to citation in text: [1] -
Ando, W.; Yagihara, T.; Tozune, S.; Imai, I.; Suzuki, J.; Toyama, T.; Nakaido, S.; Migita, T. J. Org. Chem. 1972, 37, 1721–1727. doi:10.1021/jo00976a010
Return to citation in text: [1]
| 43. | Zhou, C.-Y.; Huang, J.-S.; Che, C.-M. Synlett 2010, 2681–2700. doi:10.1055/s-0030-1259017 |
| 44. | Grohmann, M.; Buck, S.; Schäffler, L.; Maas, G. Adv. Synth. Catal. 2006, 348, 2203–2211. doi:10.1002/adsc.200606108 |
| 45. | Buck, S.; Maas, G. J. Organomet. Chem. 2006, 691, 2774–2784. doi:10.1016/j.jorganchem.2006.02.013 |
| 1. | Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; John Wiley & Sons: New York, 1998; Chapter 7. |
| 2. | Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263–309. doi:10.1021/cr00003a001 |
| 14. | Davies, H. M. L.; Crisco, L. Van T. Tetrahedron Lett. 1987, 28, 371–374. doi:10.1016/S0040-4039(00)95731-9 |
| 22. | Yufit, D. S.; Struchkov, Y. T.; Kozhushkov, S. I.; Zefirov, P. S. Russ. Chem. Bull. 1991, 40, 68–73. doi:10.1007/BF00959632 |
| 12. | Li, A.-H.; Dai, L.-X.; Aggarwal, V. K. Chem. Rev. 1997, 97, 2341–2372. doi:10.1021/cr960411r |
| 13. | Aggarwal, V. K.; Winn, C. L. Acc. Chem. Res. 2004, 37, 611–620. doi:10.1021/ar030045f |
| 23. | Igarashi, J.-e.; Watanabe, N.; Konoshita, T.; Ogata, K.; Furukawa, S. Chem. Pharm. Bull. 1990, 38, 329–335. doi:10.1248/cpb.38.329 |
| 9. | Lakeev, S. N.; Maydanova, I. O.; Galin, F. Z.; Tolstikov, G. A. Russ. Chem. Rev. 2001, 70, 655–672. doi:10.1070/RC2001v070n08ABEH000645 |
| 10. | Sweeney, J. G. Chem. Soc. Rev. 2009, 38, 1027–1038. doi:10.1039/b604828p |
| 11. | Zhang, Y.; Wang, J. Coord. Chem. Rev. 2010, 254, 941–953. doi:10.1016/j.ccr.2009.12.005 |
| 21. | Bose, A. K.; Greer, F.; Price, C. C. J. Org. Chem. 1958, 23, 1335–1338. doi:10.1021/jo01103a025 |
| 3. | Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223–270. doi:10.1021/cr950022h |
| 4. | Doyle, M. P.; Forbes, D. C. Chem. Rev. 1998, 98, 911–936. doi:10.1021/cr940066a |
| 5. | Davies, H. M. L.; Beckwith, R. E. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 6. | Wee, A. G. H. Curr. Org. Synth. 2006, 3, 499–555. doi:10.2174/157017906778699512 |
| 7. | Ferreira, V. F. Curr. Org. Chem. 2007, 11, 177–193. doi:10.2174/138527207779316462 |
| 8. | Zhang, Z.; Wang, J. Tetrahedron 2008, 64, 6577–6605. doi:10.1016/j.tet.2008.04.074 |
| 18. | Enßle, M.; Buck, S.; Werz, R.; Maas, G. ARKIVOC 2012, (iii), 149–171. |
| 19. | Padwa, A.; Dean, D. C.; Hertzog, D. L.; Nadler, W. R.; Zhi, L. Tetrahedron 1992, 48, 7565–7580. doi:10.1016/S0040-4020(01)90369-2 |
| 46. | Ando, W.; Yagihara, T.; Tozune, S.; Imai, I.; Suzuki, J.; Toyama, T.; Nakaido, S.; Migita, T. J. Org. Chem. 1972, 37, 1721–1727. doi:10.1021/jo00976a010 |
| 17. | Tolstikov, G. A.; Galin, F. Z.; Lakeev, S. N. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1989, 32, 880. |
| 14. | Davies, H. M. L.; Crisco, L. Van T. Tetrahedron Lett. 1987, 28, 371–374. doi:10.1016/S0040-4039(00)95731-9 |
| 15. | Moody, C. J.; Taylor, R. J. Tetrahedron 1990, 6501–6524. doi:10.1016/S0040-4020(01)96017-X |
| 16. | Murphy, G. K.; West, F. G. Org. Lett. 2006, 8, 4359–4361. doi:10.1021/ol061772o |
| 16. | Murphy, G. K.; West, F. G. Org. Lett. 2006, 8, 4359–4361. doi:10.1021/ol061772o |
| 15. | Moody, C. J.; Taylor, R. J. Tetrahedron 1990, 6501–6524. doi:10.1016/S0040-4020(01)96017-X |
| 15. | Moody, C. J.; Taylor, R. J. Tetrahedron 1990, 6501–6524. doi:10.1016/S0040-4020(01)96017-X |
| 20. | Nikolaev, V. V.; Krylov, I. S.; Schulze, B.; Rodina, L. L. Russ. J. Org. Chem. 2005, 41, 784–786. doi:10.1007/s11178-005-0246-3 |
| 14. | Davies, H. M. L.; Crisco, L. Van T. Tetrahedron Lett. 1987, 28, 371–374. doi:10.1016/S0040-4039(00)95731-9 |
| 24. | Standard, J. M.; Copack, B. A.; Johnson, T. K.; Przybyla, D. E.; Graham, S. R.; Steidl, R. J. J. Phys. Chem. A 2008, 112, 336–341. doi:10.1021/jp077013k |
| 18. | Enßle, M.; Buck, S.; Werz, R.; Maas, G. ARKIVOC 2012, (iii), 149–171. |
| 19. | Padwa, A.; Dean, D. C.; Hertzog, D. L.; Nadler, W. R.; Zhi, L. Tetrahedron 1992, 48, 7565–7580. doi:10.1016/S0040-4020(01)90369-2 |
| 29. | Kim, C. H.; Jang, K. P.; Choi, S. Y.; Chung, Y. K.; Lee, E. Angew. Chem., Int. Ed. 2008, 120, 4073–4075. doi:10.1002/anie.200800568 |
| 3. | Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223–270. doi:10.1021/cr950022h |
| 18. | Enßle, M.; Buck, S.; Werz, R.; Maas, G. ARKIVOC 2012, (iii), 149–171. |
| 19. | Padwa, A.; Dean, D. C.; Hertzog, D. L.; Nadler, W. R.; Zhi, L. Tetrahedron 1992, 48, 7565–7580. doi:10.1016/S0040-4020(01)90369-2 |
| 20. | Nikolaev, V. V.; Krylov, I. S.; Schulze, B.; Rodina, L. L. Russ. J. Org. Chem. 2005, 41, 784–786. doi:10.1007/s11178-005-0246-3 |
| 41. | Mehta, G.; Muthusamy, S. Tetrahedron 2002, 58, 9477–9504. doi:10.1016/S0040-4020(02)01187-0 |
| 42. | Zhou, C.-Y.; Chan, P. W. H.; Yu, W.-Y.; Che, C.-M. Synthesis 2003, 1403–1412. doi:10.1055/s-2003-40187 |
| 3. | Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223–270. doi:10.1021/cr950022h |
| 27. | Weingarten, M. D.; Prein, M.; Price, A. T.; Snyder, J. P.; Padwa, A. J. Org. Chem. 1997, 62, 2001–2010. doi:10.1021/jo962184z |
| 28. | Hodgson, D. M.; Stupple, P. A.; Pierard, F. Y. T. M.; Labande, A. H.; Johnstone, C. Chem.–Eur. J. 2001, 7, 4465–4476. doi:10.1002/1521-3765(20011015)7:20<4465::AID-CHEM4465>3.0.CO;2-W |
| 29. | Kim, C. H.; Jang, K. P.; Choi, S. Y.; Chung, Y. K.; Lee, E. Angew. Chem., Int. Ed. 2008, 120, 4073–4075. doi:10.1002/anie.200800568 |
| 30. | Mejia-Oneto, J. M.; Padwa, A. Tetrahedron Lett. 2004, 45, 9115–9118. doi:10.1016/j.tetlet.2004.10.018 |
| 31. | Hodgson, D. M.; Angrish, D.; Labande, A. H. Chem. Commun. 2006, 627–628. doi:10.1039/b515943a |
| 32. | Padwa, A.; Chughtai, M. J.; Boonsombat, J.; Rashatasakhon, P. Tetrahedron 2008, 64, 4758–4767. doi:10.1016/j.tet.2008.01.129 |
| 33. | Oh, C. H.; Lee, J. H.; Lee, S. J.; Kim, J. I.; Hong, C. S. Angew. Chem., Int. Ed. 2008, 120, 7615–7617. doi:10.1002/anie.200802425 |
| 34. | Hodgson, D. M.; Glen, R.; Redgrave, A. J. Tetrahedron: Asymmetry 2009, 20, 754–757. doi:10.1016/j.tetasy.2009.02.031 |
| 35. | Padwa, A. Tetrahedron 2011, 67, 8057–8072. doi:10.1016/j.tet.2011.07.009 |
| 36. | Padwa, A.; Hertzog, D. L.; Nadler, W. R.; Osterhout, M. H.; Price, A. T. J. Org. Chem. 1994, 59, 1418–1427. doi:10.1021/jo00085a035 |
| 37. | Padwa, A.; Heidelbaugh, T. M.; Kuethe, J. T. J. Org. Chem. 1999, 64, 2038–2049. doi:10.1021/jo982315r |
| 38. | Padwa, A.; Bur, S. K. Tetrahedron 2007, 63, 5341–5378. doi:10.1016/j.tet.2007.03.158 |
| 39. | England, D. B.; Eagan, J. M.; Merey, G.; Anac, O.; Padwa, A. Tetrahedron 2008, 64, 988–1001. doi:10.1016/j.tet.2007.10.038 |
| 40. | Li, H.; Cheng, B.; Boonnak, N.; Padwa, A. Tetrahedron 2011, 67, 9829–9836. doi:10.1016/j.tet.2011.09.118 |
| 25. | Zimmerman, H. E.; Simkin, R. D. Tetrahedron Lett. 1964, 5, 1847–1851. doi:10.1016/S0040-4039(00)75116-1 |
| 26. | Nilsen, B. J.; Undheim, K. Acta Chem. Scand. 1976, 30, 619–623. doi:10.3891/acta.chem.scand.30b-0619 |
| 3. | Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223–270. doi:10.1021/cr950022h |
| 10. | Sweeney, J. G. Chem. Soc. Rev. 2009, 38, 1027–1038. doi:10.1039/b604828p |
| 16. | Murphy, G. K.; West, F. G. Org. Lett. 2006, 8, 4359–4361. doi:10.1021/ol061772o |
© 2012 Enßle et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)