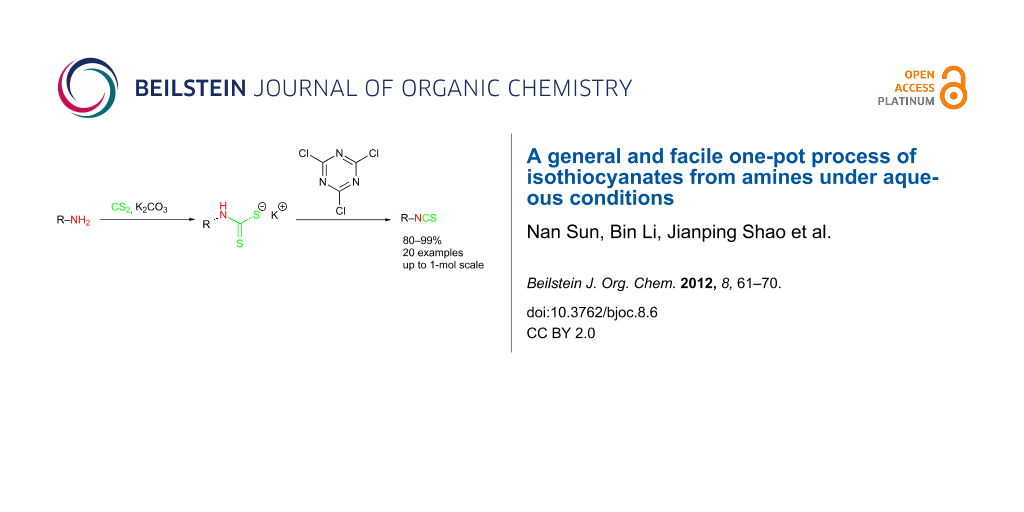
Abstract
A general and facile one-pot protocol for the preparation of a broad range of alkyl and aryl isothiocyanates has been developed from their corresponding primary amines under aqueous conditions. This synthetic process involves an in situ generation of a dithiocarbamate salt from the amine substrate by reacting with CS2 followed by elimination to form the isothiocyanate product with cyanuric acid as the desulfurylation reagent. The choice of solvent is of decisive importance for the successful formation of the dithiocarbamate salt particularly for highly electron-deficient substrates. This novel and economical method is suitable for scale-up activities.

Graphical Abstract
Introduction
Isothiocyanates are a class of heteroallenic compounds which are abundant in many cruciferous vegetables. Recently, it has been found that naturally occurring isothiocyanates play a significant role in the cancer chemopreventive activity of these plant species [1,2], and thus, many of such analogues containing the isothiocyanate motif have been synthesized for potential medical applications [3-6]. Morevoer, isothiocyanates are pivotal intermediates in organic synthesis, especially in the synthesis of various heterocyclic compounds [7,8] and unsymmetric thioureas [9-12]. Although many synthetic methods for the preparation of isothiocyanates have been reported to date [13-33], most methods suffer from the employment of highly toxic reagents such as thiophosgene and its analogs, the availability of non-commercialized reagents, or from a narrow substrate scope. Recently, a two-step approach, which was named reagent-promoted desulfurylation of dithiocarbamates strategy, has attracted much attention. In the context of this method, an amine is typically converted into a corresponding dithiocarbamate by reacting with CS2 in the presence of a base; and a subsequent desulfurylation affords the desired isothiocyanate with proper desulfurylation reagent. Although many desulfurylating reagents for this strategy were developed [34-59], most of them were efficient only for alkyl and electron-rich aryl isothiocyanates. Few efficient methods were reported for those substrates with highly electron-withdrawing groups (such as, CF3, CN, CH3CO or NO2). Therefore, the development of a more efficient method for the synthesis of isothiocyanates – particularly highly electron-deficient aryl isothiocyanates – is still a challenge in organic chemistry.
Results and Discussion
Previously, Furumoto reported the application of cyanuric chloride (2,4,6-trichloro-1,3,5-triazine, TCT) as a desulfurylation reagent in the synthesis of carbodiimides from thioureas under mild conditions [60]. In that reaction, the S-nucleophiles first reacted with TCT and then decomposed to release the product carbodiimides and by-product 2,4,6-trimercapto-1,3,5-triazine (TMT) [61]. Considering that TCT is an efficient desulfurylation reagent of thioureas to synthesize carbodiimides and that it is affordable in large scale, we speculated that it could also be effective and practical in the desulfurylation of dithiocarbamates to form isothiocyanates.
Our initial study started with aniline (1, Scheme 1) to prepare phenyl isothiocyanate (4, Scheme 1) via N-phenyl dithiocarbamate (2-1, Scheme 1) followed by desulfurylation with TCT. Because inorganic bases were more efficient during the conversion of arylamines to N-aryl dithiocarbamates in aqueous systems [54-59], we chose inorganic bases to screen the optimal reaction conditions for the one-pot synthesis of 4. Following the literature procedure, N-phenyl dithiocarbamate was generated by mixing aniline and 2.0 equiv of CS2 (based on aniline) in water with 1.0 equiv of NaOH at room temperature. After the complete disappearance of the aniline, the mixture was cooled to 0 °C and a solution of TCT (0.5 equiv) in CH2Cl2 was added. Then the biphasic mixture was stirred for 0.5 h and basified by addition of 6 N NaOH to form a clear solution. A usual work-up afforded the phenyl isothiocyanate in 61% isolated yield along with a small amount of N,N’-diphenylthiourea (Table 1, entry 1). We proposed that the overall reaction proceeded as described in Scheme 1.
![[1860-5397-8-6-i1]](/bjoc/content/inline/1860-5397-8-6-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The proposed process for the formation of N-phenyl isothiocyanate from aniline.
Scheme 1: The proposed process for the formation of N-phenyl isothiocyanate from aniline.
Table 1: Reaction of aniline with CS2 in aqueous base solutionsa.
| entry | base |
aniline/CS2/base
(equiv) |
time (h) | convb (%) |
selectb (%)
2-1:2-2 |
TCTc
(equiv) |
yieldd
(%) |
|---|---|---|---|---|---|---|---|
| 1 | NaOH | 1:2:1 | 3 | 94 | 63:35 | 0.5 | 61 |
| 2 | KOH | 1:2:1 | 3 | 93 | 72:26 | 0.5 | 63 |
| 3 | NH4OHe | 1:2:1 | 5 | 70 | 95:2 | – | – |
| 4 | Na2CO3 | 1:2:1 | 5 | 86 | 90:6 | – | – |
| 5 | NaHCO3 | 1:2:1 | 5 | 57 | 89:2 | – | – |
| 6 | K2CO3 | 1:2:1 | 5 | 86 | 97:1 | – | – |
| 7 | K2CO3 | 1:2:2 | 3 | >99 | 99:0 | 0.5 | 97 |
| 8 | K2CO3 | 1:1.2:2 | 3 | >99 | 99:0 |
0.5
0.35 |
98
89 |
aReaction conditions: 20 mmol of aniline was used in the reaction of the formation of N-phenyl dithiocarbamate at room temperature. The desulfurylation of in situ generated N-phenyl dithiocarbamate was carried out by dropwise addition of a solution of TCT in CH2Cl2 at 0 °C and then, the mixture was stirred for another 0.5 h followed by basification to pH >11 with 6 N NaOH. bDetermined by HPLC. cThe amount of TCT was based on aniline. dIsolated yield. e28% aqueous NH4OH was used.
The success observed in our preliminary study encouraged us to further optimize the conditions for better yields. Our immediate effort was to enhance the selectivity of N-phenyl dithiocarbamate over 1,3-diphenylthiourea (2-2) at the first stage. Although the formation of N-phenyl dithiocarbamate by reaction of aniline and CS2 in the presence of inorganic base under aqueous conditions has already been reported, the conversion of aniline and the selection of N-phenyl dithiocarbamate have not been disclosed before. Thus, with the help of HPLC analysis, the selectivity of N-phenyl dithiocarbamate (2-1 in Scheme 1) was first studied by testing the base effect. The results of the selectivity of 2-1 with various bases are summarized in Table 1. As can be seen from Table 1, the conversion and the selectivity were influenced by the strength of the bases. When strong bases – such as NaOH and KOH – were employed, a significant amount of 1,3-diphenylthiourea (2-2) was formed; this resulted in the low selectivity for N-phenyl dithiocarbamate (2-1) (Table 1, entry 1 and 2) despite a high conversion observed for the anilines. When weaker bases – such as NH4OH, Na2CO3 and NaHCO3 – were used, the selectivity of N-phenyl dithiocarbamate was obviously improved but the conversion of aniline decreased even at elongated reaction time (Table 1, entries 3–5). A high selectivity for 2-1 (97:1) along with high conversion (86%) was achieved (Table 1, entry 6) when K2CO3 was used. When the reaction was carried out with 2 equiv of K2CO3, a complete conversion was observed. More importantly, the selectivity of 2-1 over 2-2 was almost exclusive (Table 1, entry 7). Further studies indicated that only a slight excess of CS2 is needed for a high conversion (Table 1, entry 8).
With an efficient method for the formation of 2-1 in hand, we then continued our research on the synthesis of phenyl isothiocyanate by the desulfurylation of 2-1 with TCT. We found out that the reaction between N-phenyl dithiocarbamate and TCT proceeded rather fast, even at a lower temperature (0 °C), and 0.5 equiv of TCT was proved to be necessary for a complete conversion of N-phenyl dithiocarbamate. It turned out that the decomposition of the adduct formed between N-phenyl dithiocarbamate and TCT to release phenyl isothiocyanate was incomplete under neutral to weak basic conditions. However, when the reaction mixture was basified to pH >11 with 6 N NaOH, a complete decomposition of the adduct was observed. Moreover, under this conditions, the by-product TMT was easily soluble in water to form a clear solution and thus, convenient to the layers separation during the workup. Under optimal conditions, an overall yield of phenyl isothiocyanate from aniline was obtained up to 98% (Table 1, entry 8). It should be noted that choosing K2CO3 as the reaction base was crucial. It was found out that only the potassium salt of N-phenyl dithiocarbamate could form a clear aqueous solution, while the corresponding sodium salt was much less soluble and the ammonium salt was almost insoluble and difficult to stir. Although the isolation of intermediate 3 was not successful (Scheme 1), the fact that the formation of the side product TMT was observed strongly indicated the presence of this intermediacy.
To further study the substrate scope of this newly developed method, various amines were subjected to the treatment with CS2 in aqueous K2CO3 solution followed by desulfurylation with TCT under optimal conditions (1.2 equiv of CS2, 2.0 equiv of K2CO3 at room temperature, and 0.5 equiv of TCT in CH2Cl2 at 0 °C). The results are summarized in Table 2. As can be seen from Table 2, all tested alkylamines and electron-rich arylamines afforded their corresponding isothiocyanates in excellent yields (Table 2, entries 1–13). Compared to arylamines, alkylamines can generally be converted into their corresponding dithiocarbamates in a relative short time. This rate difference could be originated from the different basicities and their differences in water solubility. Once the dithiocarbamate was generated, the desulfurylation step using TCT proceeded smoothly for all amines. The branched alkylamines, cyclic alkylamines and benzylamines showed similar activity as linear alkylamines (Table 2, entries 1–9). The relative low yields of isopropyl isothiocyanate and tert-butyl isothiocyanate were ascribed to their volatility loss associated with isolation (Table 2, entries 1 and 3). The substituent pattern on the phenyl ring of arylamines exhibited no different activity. This also illustrated the tolerance of some steric bulk in the synthesis of isothiocyanate (Table 2, entries 11–13). Under similar reaction conditions, we then tested some electron-deficient arylamines. The difficulty in the formation of their potassium salts of dithiocarbamate was not surprising. The reaction required a slightly higher temperature and a larger excess of CS2 to complete the conversion of substrate amines. For example, 3 equiv of CS2 was needed for the conversion of 4-fluoroaniline at 40 °C in 12 h during the first step. When N-(4-fluorophenyl)dithiocarbamate was formed, 4-fluorophenyl isothiocyanate could be smoothly obtained in reasonable yield after treatment with TCT. However, under the same reaction conditions as 4-fluoroaniline, the reaction rate of 4-chloroaniline was much slower and a longer reaction time was needed. Increasing the amount of CS2 did not improve the conversion. Increasing the reaction temperature to 60 °C did improve the conversion of 4-chloroaniline but generated a lot of impurities. With 3 equiv of CS2 at 40 °C for 20 h, about 80% conversion of 4-chloroaniline with 92% selectivity of dithiocarbamate was observed. However, only 70% yield of 4-chlorophenyl isothiocyanate was obtained (Table 2, entry 15). The more electron-deficient arylamines, such as 4-trifluoromethyl- and 4-cyanoaniline, failed to convert into their corresponding isothiocyanates when the same procedure as described for 4-chloroaniline was applied.
Table 2: Preparation of isothiocyanatesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-6-i2.svg?max-width=637&scale=1.0)
|
||||
| entry | substrate | product | timeb (h) | yieldc (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-6-i3.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-6-i4.svg?max-width=637&scale=1.0)
|
1 | 85 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-6-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-6-i6.svg?max-width=637&scale=1.0)
|
1 | 94 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-6-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-6-i8.svg?max-width=637&scale=1.0)
|
1 | 80 |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-6-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-6-i10.svg?max-width=637&scale=1.0)
|
1 | 95 |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-6-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-6-i12.svg?max-width=637&scale=1.0)
|
1 | 98 |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-6-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-6-i14.svg?max-width=637&scale=1.0)
|
1 | 95 |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-6-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-6-i16.svg?max-width=637&scale=1.0)
|
1 | 99 |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-6-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-6-i18.svg?max-width=637&scale=1.0)
|
1 | 99 |
| 9 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-6-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-6-i20.svg?max-width=637&scale=1.0)
|
3 | 98 (94) |
| 10 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-6-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-6-i22.svg?max-width=637&scale=1.0)
|
3 | 86 |
| 11 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-6-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-6-i24.svg?max-width=637&scale=1.0)
|
3 | 93 |
| 12 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-6-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-6-i26.svg?max-width=637&scale=1.0)
|
3 | 95 |
| 13 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-6-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-6-i28.svg?max-width=637&scale=1.0)
|
3 | 98 |
| 14 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-8-6-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 29]](/bjoc/content/inline/1860-5397-8-6-i30.svg?max-width=637&scale=1.0)
|
12 | 81d |
| 15 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-8-6-i31.svg?max-width=637&scale=1.0)
|
![[Graphic 31]](/bjoc/content/inline/1860-5397-8-6-i32.svg?max-width=637&scale=1.0)
|
20 | 70d |
aReaction conditions: 20 mmol of amine substrate, 24 mmol of CS2, 40 mmol of K2CO3, room temperature. HPLC monitored the conversion. After the amine was totally consumed, the mixture was cooled to 0 °C, and 10 mmol of TCT in CH2Cl2 was added dropwise. The mixture was stirred for another 0.5 h and basified to pH >11 with 6 N NaOH. bThe reaction time for the first step. cIsolated yield. Data in the parenthesis is the isolated yield in 1-mol scale. d3.0 equiv of CS2 was used and the reaction temperature was 40 °C.
Moreover, to evaluate the procedure for the potential scaling-up capability, we used aniline as test substrate to scale up the synthesis in 1.0 mol scale. It became clear that the desired phenyl isothiocyanate could be conveniently isolated via vacuum distillation (Table 2, entry 9). This experiment demonstrated its potential for a larger-scale application.
The inefficiency observed for the highly electron-deficient arylamines drove us to further optimize the process. We speculated that the inefficiency for those strongly electron-deficient arylamines could be ascribed to the difficulty in the generation of dithiocarbamates between amines and CS2 in aqueous K2CO3 solution. This prompted us to seek new reaction conditions. It was reported that C2H5OH was a beneficial solvent in the conversion of electron-deficient arylamines to their corresponding dithiocarbamates with concentrated NH4OH [54]. The effect of C2H5OH in reaction media might facilitate the solubility of arylamines in water and thus, accelerate the reaction. Unfortunately, we found that CS2 would react with C2H5OH to form insoluble solid when it was charged into the reaction mixture. This solid was separated and confirmed to be potassium ethylxanthate formed between CS2 and C2H5OH in aqueous basic solution [62]. On the other hand, much better results were observed when acetonitrile, DMF or DMAc was employed. For example, with 4-chloroaniline as the substrate (Table 3), the results showed that the reaction rate of 4-chloroaniline with CS2 in the aqueous K2CO3 solution was significantly improved with acetonitrile, DMF or DMAc, and best conversion was obtained with DMF as co-solvent (Table 3). The optimized result showed that a solvent mixture of DMF/water (1:4) is suitable for electron-deficient arylamines (Table 3, entry 5).
Table 3: Preparation of 4-chlorophenyl isothiocyanate in different solventsa.
![[Graphic 32]](/bjoc/content/inline/1860-5397-8-6-i33.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent (v/v)b | time (h)c | conversion (%)d | yield (%)e |
|---|---|---|---|---|
| 1 | H2O | 20 | 56 | 50 |
| 2 | CH3CN:H2O (1:2) | 20 | 80 | 64 |
| 3 | DMAc:H2O (1:2) | 6 | 91 | 86 |
| 4 | DMF:H2O (1:2) | 6 | 98 | 91 |
| 5 | DMF:H2O (1:4) | 6 | 98 | 92 |
| 6 | DMF:H2O (1:6) | 10 | 90 | 80 |
aReaction conditions: 20 mmol of 4-chloroaniline was used in the reaction of the formation of N-(4-chlorophenyl)dithiocarbamate at 40 °C. The desulfurylation of in situ generated N-(4-chlorophenyl)dithiocarbamate was carried out by dropwise addition of a solution of TCT in CH2Cl2 at 0 °C and then the mixture was stirred another 0.5 h followed by basification to pH >11 with 6 N NaOH. DMAc = N,N-dimethyl acetamide. b20 mL of H2O used. cThe reaction time for the conversion of 4-chloroaniline to potassium N-(4-chlorophenyl)dithiocarbamate. dDetermined by HPLC. eIsolated yield.
Under optimal conditions, various electron-deficient arylamines were converted into the desired isothiocyanates in high yield and the results were listed in Table 4. With the help of DMF, more CS2 and longer reaction time, those strong electron-deficient arylamines, for examples, CF3, CN, CH3CO-substituted arylamines, could be also converted into their dithiocarbamates, which could smoothly afford high yield of their corresponding isothiocyanates by further treatment with TCT (Table 4, entries 5–7). To our surprise, in the case of 4-nitroaniline as the substrate, only 13% of 4-nitrophenyl isothiocyanate was obtained along with up to 50% of 1,4-diisothiocyanatobenzene (Table 4, entry 8). This probably resulted from the partial reduction of 4-nitroaniline to 1,4-diaminobenzene by CS2 in aqueous K2CO3 solution.
Table 4: Preparation of electron-deficient aryl isothiocyanatesa.
![[Graphic 33]](/bjoc/content/inline/1860-5397-8-6-i34.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | CS2 (equiv) | Time b (h) | Yield c (%) |
|---|---|---|---|---|---|
| 1 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-8-6-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 35]](/bjoc/content/inline/1860-5397-8-6-i36.svg?max-width=637&scale=1.0)
|
1.2 | 3 | 94 |
| 2 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-8-6-i37.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-8-6-i38.svg?max-width=637&scale=1.0)
|
2 | 6 | 92 |
| 3 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-8-6-i39.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-8-6-i40.svg?max-width=637&scale=1.0)
|
2 | 6 | 90 |
| 4 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-8-6-i41.svg?max-width=637&scale=1.0)
|
![[Graphic 41]](/bjoc/content/inline/1860-5397-8-6-i42.svg?max-width=637&scale=1.0)
|
2 | 6 | 89 |
| 5 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-8-6-i43.svg?max-width=637&scale=1.0)
|
![[Graphic 43]](/bjoc/content/inline/1860-5397-8-6-i44.svg?max-width=637&scale=1.0)
|
5 | 7 | 89 |
| 6 |
![[Graphic 44]](/bjoc/content/inline/1860-5397-8-6-i45.svg?max-width=637&scale=1.0)
|
![[Graphic 45]](/bjoc/content/inline/1860-5397-8-6-i46.svg?max-width=637&scale=1.0)
|
5 | 10 | 86 |
| 7 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-8-6-i47.svg?max-width=637&scale=1.0)
|
![[Graphic 47]](/bjoc/content/inline/1860-5397-8-6-i48.svg?max-width=637&scale=1.0)
|
5 | 10 | 91 |
| 8 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-8-6-i49.svg?max-width=637&scale=1.0)
|
![[Graphic 49]](/bjoc/content/inline/1860-5397-8-6-i50.svg?max-width=637&scale=1.0)
|
10 | 72 | 13 |
![[Graphic 50]](/bjoc/content/inline/1860-5397-8-6-i51.svg?max-width=637&scale=1.0)
|
50 | ||||
aReaction conditions: The substrate (20 mmol) was treated with excess of CS2 and 2 equiv of K2CO3 in the mixture of 5 mL of DMF and 20 mL of H2O at 40 °C. After the conversion of the substrate was complete, the mixture was cooled to 0 °C and 10 mmol of TCT in CH2Cl2 was added dropwise. The mixture was stirred for another 0.5 h and basified to pH >11 with 6 N NaOH. bThe reaction time for the conversion of amine to the corresponding dithiocarbamate. cIsolated yield.
Conclusion
In summary, we have successfully developed a facile and general method to synthesize isothiocyanates from amines. In the context of this method, the amines reacted with CS2 in aqueous K2CO3 solutions to afford dithiocarbamate intermediates, which were further desulfurylized with TCT at 0 °C to provide the corresponding isothiocyanates. The newly developed method could convert a wide range of primary alkyl and arylamines into their corresponding isothiocyanates in excellent yields and provides promise for further scale-up activities. Morevover, this method is advantageous over many other methods for the synthesis of highly electron-deficient aromatic isothiocyanates.
Experimental
General
All solvents and reagents were purchased from commercial sources and used without further purification. Melting points were measured without correction. Proton NMR spectra were recorded on a 500 MHz spectrometer in CDCl3 with tetramethylsilane (TMS) as internal standard. IR spectra were performed on a FTIR instrument. GC–MS analyses were performed in EI mode. HPLC analyses were performed using a Atlantis ODS column (150 × 4.6 mm (i.d.), 3.5 μm) with a UV detector (280 nm) at room temperature and mobile phase (60:40 CH3CN/5 mmol/L K2HPO4 water solution) with a flow rate of 0.8 mL/min. GC analyses were performed with a FID detector using a HP-5 capillary column (30 m × 0.25 mm (i.d.), 0.25 μm film thickness). The flow rate of carrier gas (N2) was 1.0 mL/min with a split ratio of 50:1. The column temperature was increased from an initial temperature of 100 to 280 °C at 10 °C/min and maintained at this temperature for 10 min. The FID detector temperature was kept at 280 °C. All operations were carried out under atmospheric conditions and all synthesized isothiocyanates were stable under the operational conditions.
Typical procedure for the preparation of alkyl and aryl isothiocyanates
To a mixture of amine (20 mmol) and K2CO3 (5.52 g, 40 mmol) in 20 mL of water 1.82 g of CS2 (24 mmol) was added dropwise in a period of 20–30 min at room temperature. After the addition was complete, the mixture was stirred for several hours until complete conversion was determined by HPLC (arylamine) or GC (alkylamine). Then, the reaction mixture was cooled to 0 °C and a solution of 1.85 g of TCT (10 mmol) in 15 mL of CH2Cl2 was added dropwise. After the addition was complete, the mixture was stirred for another 0.5 h to finish the reaction. The reaction mixture was then basified to pH >11 with 6 N NaOH to obtain a clear solution. The organic layer was separated and the aqueous phase was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and the solvent was removed in vacuo. The residual was purified by flash chromatography through a short silica column using petroleum ether as eluent.
Isopropyl isothiocyanate (CAS No: 2253-73-8, Table 2, entry 1): Yield: 85%; colorless oil; IR (neat): 2079 cm−1; 1H NMR (CDCl3) δ 1.37 (d, J = 7.0 Hz, 6H), 3.88–3.95 (m, 1H); GC–EIMS m/z: 101 [M+, 100%].
n-Butyl isothiocyanate (CAS No: 592-82-5, Table 2, entry 2): Yield: 94%; colorless oil; IR (neat): 2099 cm−1; 1H NMR (CDCl3) δ 0.94–0.97 (t, J = 7.0 Hz, 3H), 1.42–1.50 (m, 2H), 1.66–1.72 (m, 2H), 3.53 (t, J = 6.5 Hz, 2H); GC–EIMS m/z: 115 [M+, 100%].
tert-Butyl isothiocyanate (CAS No: 590-42-1, Table 2, entry 3): Yield: 80%; colorless oil; IR (neat): 2074 cm−1; 1H NMR (CDCl3) δ 1.44 (s, 9H); GC–EIMS m/z: 115 [M+, 100%].
n-Octyl isothiocyanate (CAS No: 4430-45-9, Table 2, entry 4): Yield: 95%; colorless oil; IR (neat): 2102 cm−1; 1H NMR (CDCl3) δ 0.89 (t, J = 7.0 Hz, 3H), 1.28–1.32 (m, 8H), 1.39–1.42 (m, 2H), 1.67–1.72 (m, 2H), 3.51 (t, J = 7.0 Hz, 2H); GC–EIMS m/z: 171 [M+, 0%], 115 (100%).
2-Ethyl-1-hexyl isothiocyanate (CAS No: 21663-56-9, Table 2, entry 5): Yield: 98%; colorless oil; IR (neat): 2095 cm−1; 1H NMR (CDCl3) δ 0.90–0.93 (m, 6H), 1.25–1.44 (m, 8H), 1.56–1.61 (m, 1H), 3.47–3.49 (m, 2H); GC–EIMS m/z: 171 [M+, 4%], 57 (100%).
Cyclohexyl isothiocyanate (CAS No: 1122-82-3, Table 2, entry 6): Yield: 95%; colorless oil; IR (neat): 2103 cm−1; 1H NMR (CDCl3) δ 1.38–1.90 (m, 10H), 3.67–3.71 (m, 1H); GC–EIMS m/z: 141 [M+, 90%], 55 (100%).
Benzyl isothiocyanate (CAS No: 622-78-6, Table 2, entry 7): Yield: 99%; colorless oil; IR (neat): 2091 cm−1; 1H NMR (CDCl3) δ 4.71 (s, 2H), 7.31–7.41 (m, 5H); GC–EIMS m/z: 149 [M+, 30%], 91 (100%).
1-Phenylethyl isothiocyanate (CAS No: 4478-92-6, Table 2, entry 8): Yield: 99%; colorless oil; IR (neat): 2090 cm−1; 1H NMR (CDCl3) δ 1.67 (d, J = 6.5 Hz, 3H), 4.91 (q, J = 6.5 Hz, 1H), 7.31–7.40 (m, 5H); GC–EIMS m/z: 163 [M+, 6%], 105 (100%).
Phenyl isothiocyanate (CAS No: 103-72-0, Table 2, entry 9): Yield: 98%; colorless oil; IR (neat): 2079 cm−1; 1H NMR (CDCl3) δ 7.20–7.35 (m, 5H); GC–EIMS m/z: 135 [M+, 100%].
4-Methoxyphenyl isothiocyanate (CAS No: 2284-20-0, Table 2, entry 10): Yield: 86%; colorless oil; IR (neat): 2109 cm−1; 1H NMR (CDCl3) δ 3.81 (s, 3H), 6.84–7.18 (m, 4H); GC–EIMS m/z: 165 [M+, 100%].
4-Methylphenyl isothiocyanate (CAS No: 622-59-3, Table 2, entry 11): Yield: 93%; colorless oil; IR (neat): 2100 cm−1; 1H NMR (CDCl3) δ 2.34 (s, 3H), 7.10–7.15 (m, 4H); GC–EIMS m/z: 149 [M+, 100%].
2-Methylphenyl isothiocyanate (CAS No: 614-69-7, Table 2, entry 12): Yield: 95%; colorless oil; IR (neat): 2083 cm−1; 1H NMR (CDCl3) δ 2.38 (s, 3H), 7.16–7.22 (m, 4H); GC–EIMS m/z: 149 [M+, 100%].
3-Methylphenyl isothiocyanate (CAS No: 621-30-7, Table 2, entry 13): Yield: 98%; colorless oil; IR (neat): 2106 cm−1; 1H NMR (CDCl3) δ 2.34 (s, 3H), 7.02–7.24 (m, 4H); GC–EIMS m/z: 149 [M+, 100%].
Synthetic procedure for phenyl isothiocyanate in 1-mol scale
Into a 2-L jacketed flask, 91.2 g of CS2 (1.2 mol) was dropwise added to a mixture of aniline (93.0 g, 1.0 mol) and K2CO3 (276.0 g, 2.0 mol) in 700 mL of water at room temperature within a period of 2.5 h. After the addition was complete, the mixture was stirred another 2 h. Then, the mixture was cooled to 0 °C, and a solution of 92.3 g of TCT (0.5 mol) in 450 mL of CH2Cl2 was dropwise added within 4 h. After the addition was complete, the mixture was stirred another 1 h to complete the conversion. The resulting mixture was basified to pH >11 with 250 mL of 6 N NaOH and yielded a clear solution. The organic layer was separated and the aqueous layer was extracted with 150 mL of CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent of filtrate was removed via distillation under atmospheric pressure with a 25-cm Vigreux column. The residue was vacuum distilled and the desired product fraction was collected at 72–74 °C/1 mmHg. Finally, 127.0 g of colorless liquid (94%) was obtained.
Typical procedure for the preparation of electron-deficient aryl isothiocyanates
A certain amount of CS2 and arylamine (20 mmol) was added to a solution of K2CO3 (5.52 g, 40 mmol) in 20 mL of water and 5 mL of DMF. The mixture was warmed to 40 °C for several hours to a constant conversion based on HPLC determination. The following operation was the same as the experimental procedure described above.
4-Fluorophenyl isothiocyanate (CAS No: 1544-68-9, Table 4, entry 1): Yield: 94%; colorless oil; IR (neat): 2079 cm−1; 1H NMR (CDCl3) δ 7.04–7.08 (m, 2H), 7.21–7.24 (m, 2H); GC–EIMS m/z: 153 [M+, 100%].
4-Chlorophenyl isothiocyanate (CAS No: 2131-55-7, Table 4, entry 2): Yield: 92%; white solid; mp 45–46 °C (lit. [57]: mp 44–45 °C); IR (KBr): 2037 cm−1; 1H NMR (CDCl3) δ 7.15–7.18 (m, 2H), 7.31–7.33 (m, 2H); GC–EIMS m/z: 169 [M+, 100%], 171 [M + 2, 44%].
2-Chlorophenyl isothiocyanate (CAS No: 2740-81-0, Table 4, entry 3): Yield: 90%; colorless oil; IR (neat): 2075 cm−1; 1H NMR (CDCl3) δ 7.19–7.25 (m, 3H), 7.41–7.43 (m, 1H); GC–EIMS m/z: 169 [M+, 100%], 171 [M + 2, 37%].
3-Chlorophenyl isothiocyanate (CAS No: 2392-68-9, Table 4, entry 4): Yield: 89%; colorless oil; IR (neat): 2097 cm−1; 1H NMR (CDCl3) δ 7.10–7.13 (m, 1H), 7.22–7.30 (m, 3H); GC–EIMS m/z: 169 [M+, 100%], 171 [M + 2, 34%].
4-Trifluoromethylphenyl isothiocyanate (CAS No: 1645-65-4, Table 4, entry 5): Yield: 89%; White solid; mp 40–41 °C (lit. [52]: mp 43 °C); IR (KBr): 2091 cm−1; 1H NMR (CDCl3) δ 7.31–7.34 (m, 2H), 7.62–7.63 (m, 2H); GC–EIMS m/z: 203 [M+, 100%].
4-Acetophenyl isothiocyanate (CAS No: 2131-57-9, Table 4, entry 6): Yield: 86%; white solid; mp 73–74 °C (lit. [63]: mp 75–76 °C); IR (KBr): 2125, 1680 cm−1; 1H NMR (CDCl3) δ 2.61 (s, 3H), 7.30–7.32 (m, 2H), 7.96–7.98 (m, 2H); GC–EIMS m/z: 177 [M+, 70%], 162 (100%).
4-Cyanophenyl isothiocyanate (CAS No: 2719-32-6, Table 4, entry 7): Yield: 91%; white solid; mp 121–122 °C (lit. [63]: mp 119–120 °C); IR (KBr): 2138 cm−1; 1H NMR (CDCl3) δ 7.30–7.32 (m, 2H), 7.65–7.67 (m, 2H); GC–EIMS m/z: 160 [M+, 100%].
4-Nitrophenyl isothiocyanate (CAS No: 2131-61-5, Table 4, entry 8): Yield: 13%; white solid; mp 107–108 °C (lit. [63]: mp 108–110 °C); IR (KBr): 2120 cm−1; 1H NMR (CDCl3) δ 7.36–7.38 (m, 2H), 8.26–8.27 (m, 2H); GC–EIMS m/z: 180 [M+, 100%].
1,4-Diisothiocyanatobenzene (CAS No: 4044-65-9, Table 4, entry 8): Yield: 50%; white solid; mp 129–130 °C (lit. [64]: mp 132 °C); IR (KBr): 2072 cm−1; 1H NMR (CDCl3) δ 7.22 (s, 4H); GC–EIMS m/z: 192 [M+, 100%].
Supporting Information
| Supporting Information File 1: Proton NMR and GC–MS spectra. | ||
| Format: PDF | Size: 831.3 KB | Download |
Acknowledgements
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (NSFC, 20772111, 20876149), the project of Science and Technology Department of Zhejiang Province (2009C21013) and the Natural Science Foundation of Zhejiang Province (Y407193). Hu would like to thank Dr. Tony Y. Zhang and Prof. Wenjun Tang for their helpful discussions and comments.
References
-
Fahey, J. W.; Zalcmann, A. T.; Talalay, P. Phytochemistry 2001, 56, 5–51. doi:10.1016/S0031-9422(00)00316-2
Return to citation in text: [1] -
Nakamura, Y.; Miyoshi, N. Biosci., Biotechnol., Biochem. 2010, 74, 242–255. doi:10.1271/bbb.90731
Return to citation in text: [1] -
Pedras, M. S. C.; Zheng, Q.-A.; Gadagi, R. S. Chem. Commun. 2007, 368–370. doi:10.1039/b615424g
Return to citation in text: [1] -
Munday, R.; Zhang, Y.; Munday, C. M.; Bapardekar, M. V.; Paonessa, J. D. Pharm. Res. 2008, 25, 2164–2170. doi:10.1007/s11095-008-9595-2
Return to citation in text: [1] -
Tian, X.; Huters, A. D.; Douglas, C. J.; Garg, N. K. Org. Lett. 2009, 11, 2349–2351. doi:10.1021/ol9007684
Return to citation in text: [1] -
Adsule, S.; Banerjee, S.; Ahmed, F.; Padhye, S.; Sarkar, F. H. Bioorg. Med. Chem. Lett. 2010, 20, 1247–1251. doi:10.1016/j.bmcl.2009.11.128
Return to citation in text: [1] -
Mukerjee, A. K.; Ashare, R. Chem. Rev. 1991, 91, 1–24. doi:10.1021/cr00001a001
Return to citation in text: [1] -
Sommen, G. Synlett 2004, 1323–1324. doi:10.1055/s-2004-825608
Return to citation in text: [1] -
Li, J.; Tan, Z.; Tang, S.; Hewlett, I.; Pang, R.; He, M.; He, S.; Tian, B.; Chen, K.; Yang, M. Bioorg. Med. Chem. 2009, 17, 3177–3188. doi:10.1016/j.bmc.2009.02.051
Return to citation in text: [1] -
Kang, I.-J.; Wang, L.-W.; Yeh, T.-K.; Lee, C.-C.; Lee, Y.-C.; Hsu, S.-J.; Wu, Y.-S.; Wang, J.-C.; Chao, Y.-S.; Yueh, A.; Chern, J.-H. Bioorg. Med. Chem. 2010, 18, 6414–6421. doi:10.1016/j.bmc.2010.07.002
Return to citation in text: [1] -
Peng, H.; Liang, Y.; Chen, L.; Fu, L.; Wang, H.; He, H. Bioorg. Med. Chem. Lett. 2011, 21, 1102–1104. doi:10.1016/j.bmcl.2010.12.130
Return to citation in text: [1] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r
Return to citation in text: [1] -
Toshimitsu, A.; Uemura, S.; Okano, M.; Watanabe, N. J. Org. Chem. 1983, 48, 5246–5251. doi:10.1021/jo00174a018
Return to citation in text: [1] -
Albanese, D.; Penso, M. Synthesis 1991, 1001–1002. doi:10.1055/s-1991-26629
Return to citation in text: [1] -
Shan, W. G.; Bian, G. F.; Su, W. K.; Liang, X. R. Org. Prep. Proced. Int. 2004, 36, 283–286. doi:10.1080/00304940409355967
Return to citation in text: [1] -
Kim, J. N.; Ryu, E. K. Tetrahedron Lett. 1993, 34, 8283–8284. doi:10.1016/S0040-4039(00)61411-9
Return to citation in text: [1] -
Kim, J. N.; Jung, K. S.; Lee, H. J.; Son, J. S. Tetrahedron Lett. 1997, 38, 1597–1598. doi:10.1016/S0040-4039(97)00121-4
Return to citation in text: [1] -
Adam, W.; Bargon, R. M.; Bosio, S. G.; Schenk, W. A.; Stalke, D. J. Org. Chem. 2002, 67, 7037–7041. doi:10.1021/jo026042i
Return to citation in text: [1] -
Arisawa, M.; Ashikawa, M.; Suwa, A.; Yamaguchi, M. Tetrahedron Lett. 2005, 46, 1727–1729. doi:10.1016/j.tetlet.2005.01.069
Return to citation in text: [1] -
Valette, L.; Poulain, S.; Fernandez, X.; Lizzani-Cuvelier, L. J. Sulfur Chem. 2005, 26, 155–161. doi:10.1080/17415990500070144
Return to citation in text: [1] -
Isoda, T.; Hayashi, K.; Tamai, S.; Kumagai, T.; Nagao, Y. Chem. Pharm. Bull. 2006, 54, 1616–1619. doi:10.1248/cpb.54.1616
Return to citation in text: [1] -
Zhong, B.; Al-Awar, R. S.; Shih, C.; Grimes, J. H., Jr.; Vieth, M.; Hamdouchi, C. Tetrahedron Lett. 2006, 47, 2161–2164. doi:10.1016/j.tetlet.2006.01.119
Return to citation in text: [1] -
Neely, W. J. Aust. J. Chem. 1960, 13, 341–346. doi:10.1071/CH9600341
Return to citation in text: [1] -
Bollini, M.; Casal, J. J.; Alvarez, D. E.; Boiani, L.; González, M.; Cerecetto, H.; Bruno, A. M. Bioorg. Med. Chem. 2009, 17, 1437–1444. doi:10.1016/j.bmc.2009.01.011
Return to citation in text: [1] -
Dyer, E.; Johnson, T. B. J. Am. Chem. Soc. 1932, 54, 777–787. doi:10.1021/ja01341a048
Return to citation in text: [1] -
Dyson, G. M.; Harrington, T. J. Chem. Soc. 1942, 374–375. doi:10.1039/jr9420000374
Return to citation in text: [1] -
Jochims, J. C.; Seeliger, A. Tetrahedron 1965, 21, 2611–2616. doi:10.1016/S0040-4020(01)93917-1
Return to citation in text: [1] -
Gottfried, R. Angew. Chem., Int. Ed. Engl. 1966, 5, 963–964. doi:10.1002/anie.196609632
Return to citation in text: [1] -
Larsen, C.; Steliou, K.; Harpp, D. N. J. Org. Chem. 1978, 43, 337–339. doi:10.1021/jo00396a035
Return to citation in text: [1] -
Larsen, C.; Harpp, D. N. J. Org. Chem. 1981, 46, 2465–2466. doi:10.1021/jo00325a007
Return to citation in text: [1] -
Kim, S.; Yi, K. Y. J. Org. Chem. 1986, 51, 2613–2615. doi:10.1021/jo00363a046
Return to citation in text: [1] -
Kim, S.; Yi, K. Y. Tetrahedron Lett. 1985, 26, 1661–1664. doi:10.1016/S0040-4039(00)98578-2
Return to citation in text: [1] -
Grayson, J. I. Org. Process Res. Dev. 1997, 1, 240–246. doi:10.1021/op970002c
Return to citation in text: [1] -
Hodgkins, J. E.; Ettlinger, M. G. J. Org. Chem. 1956, 21, 404–405. doi:10.1021/jo01110a006
Return to citation in text: [1] -
Hodgkins, J. E.; Reeves, W. P. J. Org. Chem. 1964, 29, 3098–3099. doi:10.1021/jo01033a524
Return to citation in text: [1] -
Shibanuma, T.; Shiono, M.; Mukaiyama, T. Chem. Lett. 1977, 6, 573–574. doi:10.1246/cl.1977.573
Return to citation in text: [1] -
Kondo, K.; Komamura, C.; Murakami, M.; Takemoto, K. Synth. Commun. 1985, 15, 171–177. doi:10.1080/00397918508063784
Return to citation in text: [1] -
Yamamoto, T.; Terada, A.; Muramatsu, T.; Ikeda, K. Org. Prep. Proced. Int. 1994, 26, 555–557. doi:10.1080/00304949409458055
Return to citation in text: [1] -
Boas, U.; Jakobsen, M. H. J. Chem. Soc., Chem. Commun. 1995, 1995–1996. doi:10.1039/C39950001995
Return to citation in text: [1] -
Boas, U.; Pedersen, B.; Christensen, J. B. Synth. Commun. 1998, 28, 1223–1231. doi:10.1080/00397919808005964
Return to citation in text: [1] -
Boas, U.; Gertz, H.; Christensen, J. B.; Heegaard, P. M. H. Tetrahedron Lett. 2004, 45, 269–272. doi:10.1016/j.tetlet.2003.10.182
Return to citation in text: [1] -
Li, G.; Tajima, H.; Ohtani, T. J. Org. Chem. 1997, 62, 4539–4540. doi:10.1021/jo970100w
Return to citation in text: [1] -
Isobe, T. J. Org. Chem. 1999, 64, 6984–6988. doi:10.1021/jo990210y
Return to citation in text: [1] -
Bian, G.; Shan, W.; Su, W. J. Chem. Res. 2005, 585–586. doi:10.3184/030823405774308862
Return to citation in text: [1] -
Chaskar, A. C.; Yewale, S.; Bhagat, R.; Langi, B. P. Synth. Commun. 2008, 38, 1972–1975. doi:10.1080/00397910801997744
Return to citation in text: [1] -
Wong, R.; Dolman, S. J. J. Org. Chem. 2007, 72, 3969–3971. doi:10.1021/jo070246n
Return to citation in text: [1] -
Bian, G.; Qiu, H.; Jiang, J.; Wu, J.; Lai, G. Phosphorus, Sulfur Silicon Relat. Elem. 2007, 182, 503–508. doi:10.1080/10426500600977379
Return to citation in text: [1] -
Ghosh, H.; Yella, R.; Nath, J.; Patel, B. K. Eur. J. Org. Chem. 2008, 6189–6196. doi:10.1002/ejoc.200800901
Return to citation in text: [1] -
Munch, H.; Hansen, J. S.; Pittelkow, M.; Christensen, J. B.; Boas, U. Tetrahedron Lett. 2008, 49, 3117–3119. doi:10.1016/j.tetlet.2008.03.045
Return to citation in text: [1] -
Chaskar, A. C.; Bandgar, B. P.; Modhave, R. K.; Patil, A. B.; Yewale, S. Synth. Commun. 2009, 39, 992–1001. doi:10.1080/00397910802448481
Return to citation in text: [1] -
Nath, J.; Ghosh, H.; Yella, R.; Patel, B. K. Eur. J. Org. Chem. 2009, 1849–1851. doi:10.1002/ejoc.200801270
Return to citation in text: [1] -
Jamir, L.; Ali, A. R.; Ghosh, H.; Chipem, F. A. S.; Patel, B. K. Org. Biomol. Chem. 2010, 8, 1674–1678. doi:10.1039/b923336a
Return to citation in text: [1] [2] -
Yella, R.; Ghosh, H.; Murru, S.; Sahoo, S. K.; Patel, B. K. Synth. Commun. 2010, 40, 2083–2096. doi:10.1080/00397910903219476
Return to citation in text: [1] -
Dains, F. B.; Brewster, R. G.; Olander, C. P. Organic Syntheses; John Wiley & Sons: New York, NY, 1941; Vol. 1, p 447.
Return to citation in text: [1] [2] [3] -
Moore, M. L.; Crossley, F. S. Organic Syntheses; John Wiley & Sons: New York, NY, 1955; Vol. 3, p 599.
Return to citation in text: [1] [2] -
Li, Z.; Qian, X.; Liu, Z.; Li, Z.; Song, G. Org. Prep. Proced. Int. 2000, 32, 571–573. doi:10.1080/00304940009355953
Return to citation in text: [1] [2] -
van der Kerk, G. J. M.; Pluygers, C. W.; de Vries, G. Organic Syntheses; John Wiley & Sons: New York, NY, 1973; Vol. 5, p 223.
Return to citation in text: [1] [2] [3] -
Heusenstamm, G. G.; Schreyer, G.; Vanheertum, R. U.S. Patent 4,089,887, May 16, 1978.
Return to citation in text: [1] [2] -
Mesheram, H. M.; Dale, S.; Yadav, J. S. Tetrahedron Lett. 1997, 38, 8743–8744. doi:10.1016/S0040-4039(97)10158-7
Return to citation in text: [1] [2] -
Furumoto, S. Nippon Kagaku Zasshi 1971, 92, 1005–1007. doi:10.1246/nikkashi1948.92.1005
Return to citation in text: [1] -
Henke, K. R.; Hutchison, A. R.; Krepps, M. K.; Parkin, S.; Atwood, D. A. Inorg. Chem. 2001, 40, 4443–4447. doi:10.1021/ic0103188
Return to citation in text: [1] -
Xiao, W.-J.; Alper, H. J. Org. Chem. 1999, 64, 9646–9652. doi:10.1021/jo9913098
Return to citation in text: [1] -
Sayigh, A. A. R.; Ulrich, H.; Potts, J. S. J. Org. Chem. 1965, 30, 2465–2466. doi:10.1021/jo01018a511
Return to citation in text: [1] [2] [3] -
Lieber, E.; Slutkin, R. J. Org. Chem. 1962, 27, 2214–2217. doi:10.1021/jo01053a509
Return to citation in text: [1]
| 64. | Lieber, E.; Slutkin, R. J. Org. Chem. 1962, 27, 2214–2217. doi:10.1021/jo01053a509 |
| 1. | Fahey, J. W.; Zalcmann, A. T.; Talalay, P. Phytochemistry 2001, 56, 5–51. doi:10.1016/S0031-9422(00)00316-2 |
| 2. | Nakamura, Y.; Miyoshi, N. Biosci., Biotechnol., Biochem. 2010, 74, 242–255. doi:10.1271/bbb.90731 |
| 13. | Toshimitsu, A.; Uemura, S.; Okano, M.; Watanabe, N. J. Org. Chem. 1983, 48, 5246–5251. doi:10.1021/jo00174a018 |
| 14. | Albanese, D.; Penso, M. Synthesis 1991, 1001–1002. doi:10.1055/s-1991-26629 |
| 15. | Shan, W. G.; Bian, G. F.; Su, W. K.; Liang, X. R. Org. Prep. Proced. Int. 2004, 36, 283–286. doi:10.1080/00304940409355967 |
| 16. | Kim, J. N.; Ryu, E. K. Tetrahedron Lett. 1993, 34, 8283–8284. doi:10.1016/S0040-4039(00)61411-9 |
| 17. | Kim, J. N.; Jung, K. S.; Lee, H. J.; Son, J. S. Tetrahedron Lett. 1997, 38, 1597–1598. doi:10.1016/S0040-4039(97)00121-4 |
| 18. | Adam, W.; Bargon, R. M.; Bosio, S. G.; Schenk, W. A.; Stalke, D. J. Org. Chem. 2002, 67, 7037–7041. doi:10.1021/jo026042i |
| 19. | Arisawa, M.; Ashikawa, M.; Suwa, A.; Yamaguchi, M. Tetrahedron Lett. 2005, 46, 1727–1729. doi:10.1016/j.tetlet.2005.01.069 |
| 20. | Valette, L.; Poulain, S.; Fernandez, X.; Lizzani-Cuvelier, L. J. Sulfur Chem. 2005, 26, 155–161. doi:10.1080/17415990500070144 |
| 21. | Isoda, T.; Hayashi, K.; Tamai, S.; Kumagai, T.; Nagao, Y. Chem. Pharm. Bull. 2006, 54, 1616–1619. doi:10.1248/cpb.54.1616 |
| 22. | Zhong, B.; Al-Awar, R. S.; Shih, C.; Grimes, J. H., Jr.; Vieth, M.; Hamdouchi, C. Tetrahedron Lett. 2006, 47, 2161–2164. doi:10.1016/j.tetlet.2006.01.119 |
| 23. | Neely, W. J. Aust. J. Chem. 1960, 13, 341–346. doi:10.1071/CH9600341 |
| 24. | Bollini, M.; Casal, J. J.; Alvarez, D. E.; Boiani, L.; González, M.; Cerecetto, H.; Bruno, A. M. Bioorg. Med. Chem. 2009, 17, 1437–1444. doi:10.1016/j.bmc.2009.01.011 |
| 25. | Dyer, E.; Johnson, T. B. J. Am. Chem. Soc. 1932, 54, 777–787. doi:10.1021/ja01341a048 |
| 26. | Dyson, G. M.; Harrington, T. J. Chem. Soc. 1942, 374–375. doi:10.1039/jr9420000374 |
| 27. | Jochims, J. C.; Seeliger, A. Tetrahedron 1965, 21, 2611–2616. doi:10.1016/S0040-4020(01)93917-1 |
| 28. | Gottfried, R. Angew. Chem., Int. Ed. Engl. 1966, 5, 963–964. doi:10.1002/anie.196609632 |
| 29. | Larsen, C.; Steliou, K.; Harpp, D. N. J. Org. Chem. 1978, 43, 337–339. doi:10.1021/jo00396a035 |
| 30. | Larsen, C.; Harpp, D. N. J. Org. Chem. 1981, 46, 2465–2466. doi:10.1021/jo00325a007 |
| 31. | Kim, S.; Yi, K. Y. J. Org. Chem. 1986, 51, 2613–2615. doi:10.1021/jo00363a046 |
| 32. | Kim, S.; Yi, K. Y. Tetrahedron Lett. 1985, 26, 1661–1664. doi:10.1016/S0040-4039(00)98578-2 |
| 33. | Grayson, J. I. Org. Process Res. Dev. 1997, 1, 240–246. doi:10.1021/op970002c |
| 63. | Sayigh, A. A. R.; Ulrich, H.; Potts, J. S. J. Org. Chem. 1965, 30, 2465–2466. doi:10.1021/jo01018a511 |
| 9. | Li, J.; Tan, Z.; Tang, S.; Hewlett, I.; Pang, R.; He, M.; He, S.; Tian, B.; Chen, K.; Yang, M. Bioorg. Med. Chem. 2009, 17, 3177–3188. doi:10.1016/j.bmc.2009.02.051 |
| 10. | Kang, I.-J.; Wang, L.-W.; Yeh, T.-K.; Lee, C.-C.; Lee, Y.-C.; Hsu, S.-J.; Wu, Y.-S.; Wang, J.-C.; Chao, Y.-S.; Yueh, A.; Chern, J.-H. Bioorg. Med. Chem. 2010, 18, 6414–6421. doi:10.1016/j.bmc.2010.07.002 |
| 11. | Peng, H.; Liang, Y.; Chen, L.; Fu, L.; Wang, H.; He, H. Bioorg. Med. Chem. Lett. 2011, 21, 1102–1104. doi:10.1016/j.bmcl.2010.12.130 |
| 12. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 63. | Sayigh, A. A. R.; Ulrich, H.; Potts, J. S. J. Org. Chem. 1965, 30, 2465–2466. doi:10.1021/jo01018a511 |
| 7. | Mukerjee, A. K.; Ashare, R. Chem. Rev. 1991, 91, 1–24. doi:10.1021/cr00001a001 |
| 8. | Sommen, G. Synlett 2004, 1323–1324. doi:10.1055/s-2004-825608 |
| 52. | Jamir, L.; Ali, A. R.; Ghosh, H.; Chipem, F. A. S.; Patel, B. K. Org. Biomol. Chem. 2010, 8, 1674–1678. doi:10.1039/b923336a |
| 3. | Pedras, M. S. C.; Zheng, Q.-A.; Gadagi, R. S. Chem. Commun. 2007, 368–370. doi:10.1039/b615424g |
| 4. | Munday, R.; Zhang, Y.; Munday, C. M.; Bapardekar, M. V.; Paonessa, J. D. Pharm. Res. 2008, 25, 2164–2170. doi:10.1007/s11095-008-9595-2 |
| 5. | Tian, X.; Huters, A. D.; Douglas, C. J.; Garg, N. K. Org. Lett. 2009, 11, 2349–2351. doi:10.1021/ol9007684 |
| 6. | Adsule, S.; Banerjee, S.; Ahmed, F.; Padhye, S.; Sarkar, F. H. Bioorg. Med. Chem. Lett. 2010, 20, 1247–1251. doi:10.1016/j.bmcl.2009.11.128 |
| 63. | Sayigh, A. A. R.; Ulrich, H.; Potts, J. S. J. Org. Chem. 1965, 30, 2465–2466. doi:10.1021/jo01018a511 |
| 54. | Dains, F. B.; Brewster, R. G.; Olander, C. P. Organic Syntheses; John Wiley & Sons: New York, NY, 1941; Vol. 1, p 447. |
| 55. | Moore, M. L.; Crossley, F. S. Organic Syntheses; John Wiley & Sons: New York, NY, 1955; Vol. 3, p 599. |
| 56. | Li, Z.; Qian, X.; Liu, Z.; Li, Z.; Song, G. Org. Prep. Proced. Int. 2000, 32, 571–573. doi:10.1080/00304940009355953 |
| 57. | van der Kerk, G. J. M.; Pluygers, C. W.; de Vries, G. Organic Syntheses; John Wiley & Sons: New York, NY, 1973; Vol. 5, p 223. |
| 58. | Heusenstamm, G. G.; Schreyer, G.; Vanheertum, R. U.S. Patent 4,089,887, May 16, 1978. |
| 59. | Mesheram, H. M.; Dale, S.; Yadav, J. S. Tetrahedron Lett. 1997, 38, 8743–8744. doi:10.1016/S0040-4039(97)10158-7 |
| 62. | Xiao, W.-J.; Alper, H. J. Org. Chem. 1999, 64, 9646–9652. doi:10.1021/jo9913098 |
| 61. | Henke, K. R.; Hutchison, A. R.; Krepps, M. K.; Parkin, S.; Atwood, D. A. Inorg. Chem. 2001, 40, 4443–4447. doi:10.1021/ic0103188 |
| 57. | van der Kerk, G. J. M.; Pluygers, C. W.; de Vries, G. Organic Syntheses; John Wiley & Sons: New York, NY, 1973; Vol. 5, p 223. |
| 60. | Furumoto, S. Nippon Kagaku Zasshi 1971, 92, 1005–1007. doi:10.1246/nikkashi1948.92.1005 |
| 34. | Hodgkins, J. E.; Ettlinger, M. G. J. Org. Chem. 1956, 21, 404–405. doi:10.1021/jo01110a006 |
| 35. | Hodgkins, J. E.; Reeves, W. P. J. Org. Chem. 1964, 29, 3098–3099. doi:10.1021/jo01033a524 |
| 36. | Shibanuma, T.; Shiono, M.; Mukaiyama, T. Chem. Lett. 1977, 6, 573–574. doi:10.1246/cl.1977.573 |
| 37. | Kondo, K.; Komamura, C.; Murakami, M.; Takemoto, K. Synth. Commun. 1985, 15, 171–177. doi:10.1080/00397918508063784 |
| 38. | Yamamoto, T.; Terada, A.; Muramatsu, T.; Ikeda, K. Org. Prep. Proced. Int. 1994, 26, 555–557. doi:10.1080/00304949409458055 |
| 39. | Boas, U.; Jakobsen, M. H. J. Chem. Soc., Chem. Commun. 1995, 1995–1996. doi:10.1039/C39950001995 |
| 40. | Boas, U.; Pedersen, B.; Christensen, J. B. Synth. Commun. 1998, 28, 1223–1231. doi:10.1080/00397919808005964 |
| 41. | Boas, U.; Gertz, H.; Christensen, J. B.; Heegaard, P. M. H. Tetrahedron Lett. 2004, 45, 269–272. doi:10.1016/j.tetlet.2003.10.182 |
| 42. | Li, G.; Tajima, H.; Ohtani, T. J. Org. Chem. 1997, 62, 4539–4540. doi:10.1021/jo970100w |
| 43. | Isobe, T. J. Org. Chem. 1999, 64, 6984–6988. doi:10.1021/jo990210y |
| 44. | Bian, G.; Shan, W.; Su, W. J. Chem. Res. 2005, 585–586. doi:10.3184/030823405774308862 |
| 45. | Chaskar, A. C.; Yewale, S.; Bhagat, R.; Langi, B. P. Synth. Commun. 2008, 38, 1972–1975. doi:10.1080/00397910801997744 |
| 46. | Wong, R.; Dolman, S. J. J. Org. Chem. 2007, 72, 3969–3971. doi:10.1021/jo070246n |
| 47. | Bian, G.; Qiu, H.; Jiang, J.; Wu, J.; Lai, G. Phosphorus, Sulfur Silicon Relat. Elem. 2007, 182, 503–508. doi:10.1080/10426500600977379 |
| 48. | Ghosh, H.; Yella, R.; Nath, J.; Patel, B. K. Eur. J. Org. Chem. 2008, 6189–6196. doi:10.1002/ejoc.200800901 |
| 49. | Munch, H.; Hansen, J. S.; Pittelkow, M.; Christensen, J. B.; Boas, U. Tetrahedron Lett. 2008, 49, 3117–3119. doi:10.1016/j.tetlet.2008.03.045 |
| 50. | Chaskar, A. C.; Bandgar, B. P.; Modhave, R. K.; Patil, A. B.; Yewale, S. Synth. Commun. 2009, 39, 992–1001. doi:10.1080/00397910802448481 |
| 51. | Nath, J.; Ghosh, H.; Yella, R.; Patel, B. K. Eur. J. Org. Chem. 2009, 1849–1851. doi:10.1002/ejoc.200801270 |
| 52. | Jamir, L.; Ali, A. R.; Ghosh, H.; Chipem, F. A. S.; Patel, B. K. Org. Biomol. Chem. 2010, 8, 1674–1678. doi:10.1039/b923336a |
| 53. | Yella, R.; Ghosh, H.; Murru, S.; Sahoo, S. K.; Patel, B. K. Synth. Commun. 2010, 40, 2083–2096. doi:10.1080/00397910903219476 |
| 54. | Dains, F. B.; Brewster, R. G.; Olander, C. P. Organic Syntheses; John Wiley & Sons: New York, NY, 1941; Vol. 1, p 447. |
| 55. | Moore, M. L.; Crossley, F. S. Organic Syntheses; John Wiley & Sons: New York, NY, 1955; Vol. 3, p 599. |
| 56. | Li, Z.; Qian, X.; Liu, Z.; Li, Z.; Song, G. Org. Prep. Proced. Int. 2000, 32, 571–573. doi:10.1080/00304940009355953 |
| 57. | van der Kerk, G. J. M.; Pluygers, C. W.; de Vries, G. Organic Syntheses; John Wiley & Sons: New York, NY, 1973; Vol. 5, p 223. |
| 58. | Heusenstamm, G. G.; Schreyer, G.; Vanheertum, R. U.S. Patent 4,089,887, May 16, 1978. |
| 59. | Mesheram, H. M.; Dale, S.; Yadav, J. S. Tetrahedron Lett. 1997, 38, 8743–8744. doi:10.1016/S0040-4039(97)10158-7 |
| 54. | Dains, F. B.; Brewster, R. G.; Olander, C. P. Organic Syntheses; John Wiley & Sons: New York, NY, 1941; Vol. 1, p 447. |
© 2012 Sun et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)