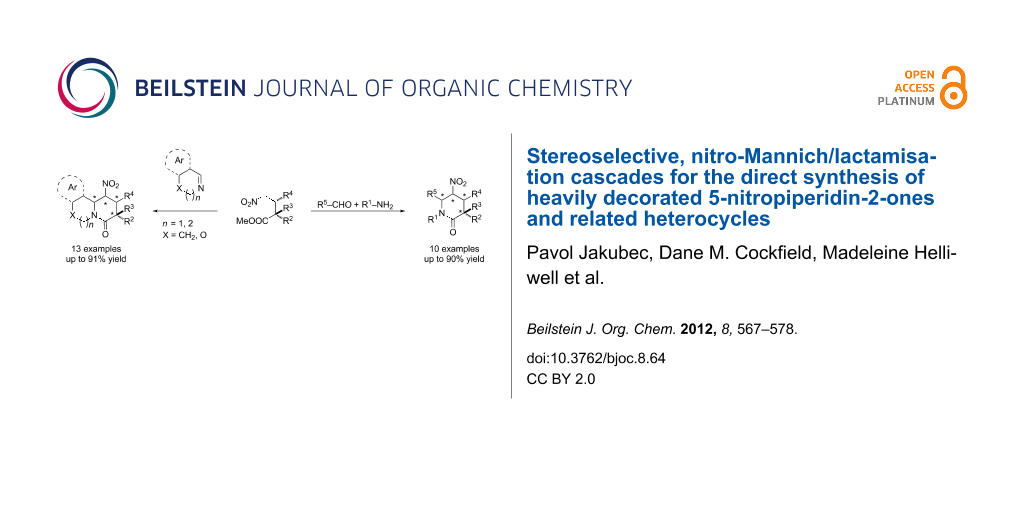
Abstract
A versatile nitro-Mannich/lactamisation cascade for the direct stereoselective synthesis of heavily decorated 5-nitropiperidin-2-ones and related heterocycles has been developed. A highly enantioenriched substituted 5-nitropiperidin-2-one was synthesised in a four component one-pot reaction combining an enantioselective organocatalytic Michael addition with the diastereoselective nitro-Mannich/lactamisation cascade. Protodenitration and chemoselective reductive manipulation of the heterocycles was used to install contiguous and fully substituted stereocentres in the synthesis of substituted piperidines.

Graphical Abstract
Introduction
The piperidine ring is a common motif found in many biologically active natural products and drugs. The structures of these compounds range from the architecturally complex polycyclic ring systems, such as those found in the alkaloids haliclonacyclamine F [1], manzamine A [2-6], and reserpine [7,8] (Figure 1), to relatively simple piperidines found in pharmaceutical compounds, such as paroxetine [9,10] and alvimopan [11].
![[1860-5397-8-64-1]](/bjoc/content/figures/1860-5397-8-64-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active natural products and drugs containing the piperidine ring.
Figure 1: Biologically active natural products and drugs containing the piperidine ring.
The abundance of this motif in desirable targets has led to considerable interest from the synthetic community [12-19]. Common synthetic approaches to incorporate this motif include nucleophilic additions to pyridine rings and further manipulation [20-25], intramolecular iminium ion cyclisation [26-29], reduction of unsaturated heterocycles [30-32], ring closure via intramolecular nucleophilic substitution [33-37], cascade reactions of enamines/imines and aldehydes [38-41], and ring-closing metathesis followed by hydrogenation [42-48]. Arguably the most general route employs cycloadditions and subsequent manipulation of the partially unsaturated ring system [49-53]. We believed a powerful entry to piperidine rings and related heterocyclic structures could employ a nitro-Mannich/lactamisation cascade of γ-nitro ester starting materials with imines (cyclic or acyclic, preformed or formed in situ) as a key step. Not only could this approach allow the rapid generation of structural complexity, but the products would be amenable to further synthetic transformations. Furthermore, the γ-nitro ester starting materials are accessible in an enantioenriched form by using an organocatalytic Michael addition methodology, which was developed by our group and others [54-59]. In pursuit of this we have successfully harnessed the power of the nitro-Mannich/lactamisation cascade in a formal synthesis of (3S,4R)-paroxetine [60], in the construction of architecturally complex polycyclic alkaloid structures [61] and more recently as a key complexity building step in the total synthesis of nakadomarin A [62-65]. Herein we wish to report our full findings in this synthetically powerful cyclisation cascade.
The first example of a simple nitro-Mannich/lactamisation cascade was reported independently by Mühlstädt and Jain in the mid-1970s [66,67]. The condensation of methyl 4-nitrobutanoate 6 (Scheme 1; R2 = R3 = R4 = H) with aromatic aldehydes 3 (R5 = Ar) and ammonium acetate provided access to simple 6-aryl-substituted 5-nitropiperidin-2-ones 1 (R1 = R2 = R3 = R4 = H, R5 = Ar). The power of this transformation was not immediately recognised and only in the last two decades has the cascade been successfully applied to the synthesis of simple biologically active compounds and their precursors, such as (±)-CP-99,994 [68,69], inhibitors of farnesyltransferase [70,71], selective dipeptidyl peptidase IV inhibitors [72,73], and functionalised bispidines [74]. Very recently, a related cascade inspired by the original work of Jain incorporating C–C bond formation was accomplished through a nitro-Mannich reaction [75-80] of nitro carbonyl compounds with imines, followed by ring-closure condensation [81-83]. Despite improvements of, and developments to, the nitro-Mannich/lactamisation cascade during the last few decades, we recognised, that further enhancement of the method was necessary to transform it into a general synthetic tool of use in both medicinal chemistry and natural-product synthesis.
![[1860-5397-8-64-i1]](/bjoc/content/inline/1860-5397-8-64-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A general strategy to 5-nitropiperidin-2-ones and related heterocycles.
Scheme 1: A general strategy to 5-nitropiperidin-2-ones and related heterocycles.
Results and Discussion
To allow us to further explore the nitro-Mannich/lactamisation cascade, a range of Michael adducts 6a–e were synthesised on a gram scale by the reaction of active methylene or methine carbon acids with nitro olefins in the presence of DABCO (20–30 mol %) in THF (Scheme 2). Where diastereoisomers were created in the Michael addition step and stereocontrol was poor, the diastereomeric mixtures were recrystallised to afford single diastereomers 6a, b, e. The relative stereochemistry of the major diastereomer 6e was assigned unambiguously by single-crystal X-ray analysis.
![[1860-5397-8-64-i2]](/bjoc/content/inline/1860-5397-8-64-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of Michael adduct model substrates for the nitro-Mannich/lactamisation cascade.
Scheme 2: The synthesis of Michael adduct model substrates for the nitro-Mannich/lactamisation cascade.
With a range of suitable test substrates in hand, formaldehyde-derived imines were then investigated in the nitro-Mannich/lactamisation reaction. Aqueous formaldehyde (3a) and allylamine (4a) were added to a methanol solution of lactam 6a and the mixture heated under reflux for 4 hours until judged to be complete by TLC. Pleasingly, the desired δ-lactam product 1a was isolated in 90% yield as a single diasteromer (Scheme 3) [84]. Under identical reaction conditions the other Michael adducts, lactone 6b and ester 6c, provided moderate yields of the desired δ-lactams 1b and 1c as single diastereoisomers in both cases. The diastereoselectivity in the latter case is notable, as the quaternary stereogenic centre is created in the lactamisation step. The relative stereochemical configurations of 1a–c were established by 1H NMR spectroscopic analysis. For more details on the elucidation of the relative configuration see [61] and Supporting Information File 1. To incorporate substituents at the 6 position of the piperidine ring in 1, imines derived from aldehydes other than formaldehyde were required in the reaction. Thus acetaldehyde (3b), anisaldehyde (3c) and glyoxylic acid (3d) were chosen as representative aliphatic, aromatic and functionalised aldehydes, respectively, and reacted with Michael adducts 6a and 6d under the conditions described above with allylamine. High diastereoselectivities were observed in each case and the reaction products 1d–g were obtained in moderate to good yields (50–74%). The relative stereochemistry of 1g was assigned unambiguously by single-crystal X-ray analysis. Similarly, variation at position 1 required the use of an alternative amine for in situ imine formation. Thus replacement of allylamine (4a) with benzylamine (4b) in the reaction afforded the desired product 1h in good yield and as a single diastereoisomer (Scheme 3).
![[1860-5397-8-64-i3]](/bjoc/content/inline/1860-5397-8-64-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Nitro-Mannich/lactamisation cascade with in situ formed imines.
Scheme 3: Nitro-Mannich/lactamisation cascade with in situ formed imines.
The use of substrate 6e allowed us to investigate further variations at positions 1 and 4; piperidin-2-ones 1i and 1j were formed as single diastereoisomers in good yields (82% and 75%) when nitro-Mannich/lactamisation cascades were carried out with formaldehyde (3a) and butylamine (4c) or hept-5-yn-1-amine (4d), respectively. To extend the cascade methodology to the potential construction of architecturally complex piperidine-ring-containing polycyclic natural products, the successful employment of preformed cyclic imines was required.
Accordingly, the imine 5a (Figure 2) was synthesised from commercially available 2-phenylethylamine [85] and reacted with the chromatographically inseparable mixture of diastereomeric Michael adducts 6a and 6a’’, under slightly modified conditions (water was used instead of MeOH as the solvent). Pleasingly the reaction proceeded smoothly and only two, 2a and 2a’’, of the possible eight diastereoisomeric tetracyclic compounds were obtained in good combined yield (70%, Scheme 4). Chromatographic separation followed by single-crystal X-ray diffraction studies of both isomers allowed unambiguous determination of the relative stereochemical configurations in each case. For more details of the elucidation of the relative configuration see Supporting Information File 1. The products were epimeric only at the quaternary centre and therefore both new stereogenic centres were created with high stereocontrol in each case.
![[1860-5397-8-64-2]](/bjoc/content/figures/1860-5397-8-64-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyclic imines employed in nitro-Mannich/lactamisation cascade.
Figure 2: Cyclic imines employed in nitro-Mannich/lactamisation cascade.
![[1860-5397-8-64-i4]](/bjoc/content/inline/1860-5397-8-64-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Nitro-Mannich/lactamisation cascade of diastereomeric Michael adducts 6a, 6a’’ with cyclic imine 5a.
Scheme 4: Nitro-Mannich/lactamisation cascade of diastereomeric Michael adducts 6a, 6a’’ with cyclic imine 5a....
Imines 5a–5i [61,86-94], chosen so as to afford common target motifs in the products [95-102], were synthesised and reacted with diastereomerically pure Michael adduct 6a and Michael adduct 6d following the conditions described above. Employing the optimal reaction conditions, products 2a–2l, which possess 4,5-trans relative stereochemistry, were formed in moderate to good yields and with high diastereoselectivities as described in our previous work (Scheme 5) [61].
![[1860-5397-8-64-i5]](/bjoc/content/inline/1860-5397-8-64-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Nitro-Mannich/lactamisation cascade with cyclic imines. aDiastereomeric ratio in a crude reaction mixture, bH2O/MeOH 1:1 mixture used as a solvent, cminor diastereomer 2m isolated in 5% yield.
Scheme 5: Nitro-Mannich/lactamisation cascade with cyclic imines. aDiastereomeric ratio in a crude reaction m...
Interestingly, however, when diastereomerically pure Michael adduct 6d was reacted with imine 5e, the nitropiperidinone 2m’, possessing 4,5-cis relative stereochemistry [103], was isolated in 70% yield as a single diastereomer (Scheme 5). This one exceptional case together with the generally high diastereocontrol in the formation of piperidinones 1a–j and 2a–l is interesting and worthy of further commentary. With the knowledge that the retro-Michael reaction does not occur under standard reaction conditions (Scheme 4) and assuming that the final step of the cascade (the δ-lactam ring formation) is irreversible, there are at least three possible explanations for the high diastereocontrol in the formation of 1a–j and 2a–l:
- The first is that the nitro-Mannich step is highly diastereoselective and lactamisation occurs subsequently without any effect on the stereochemical outcome of the cascade.
- The second is that the nitro-Mannich reaction [78-80] is fast and reversible (but not necessarily stereoselective), and only one of the diastereomeric nitro-Mannich products preferentially cyclises in the irreversible lactamisation step to the (likely) most thermodynamically stable product (Scheme 6, Path A).
- The third is similar to the second, but the two direct nitro-Mannich products A and B with the observed configurations at the 6 position preferentially lactamise, and there is a postcyclisation epimerisation at the stereogenic carbon bearing the nitro group allowing equilibration to the (likely) most thermodynamically stable product (thermodynamic control, Path B) or a crystallisation-induced diastereoselectivity to give products 2 or 2’ with the nitro group occupying an axial or equatorial position, respectively [104-108] (Scheme 6, Path B/B’).
![[1860-5397-8-64-i6]](/bjoc/content/inline/1860-5397-8-64-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible explanations for the observed high stereoselectivities in the nitro-Mannich/lactamisation cascade.
Scheme 6: Possible explanations for the observed high stereoselectivities in the nitro-Mannich/lactamisation ...
A further scrutiny of each hypothesis was, unfortunately, hampered by our failure to isolate or identify in situ the direct nitro-Mannich products from the reaction mixtures or to prepare them separately using standard procedures for a nitro-Mannich reaction with imines [109]. The first hypothesis, however, was not supported by the low diastereoselectivity in the formation of 2j, in which presumably the relatively fast irreversible cyclisation outcompetes the equilibration processes. Considering the relatively broad range of imines and Michael adducts involved in the stereoselective cascade, we believe that the second or third explanations are the most plausible and that the observed diastereoselectivities in the formation of products 1a–j, 2a–l can be explained by following either Path A or B (Scheme 6).
The formation of product 2m’ with its exceptional 4,5-cis relative stereochemistry, can be explained by following path B’ (Scheme 6). In this case the observed diastereoselectivity is believed to be driven by preferential crystallisation of the 4,5-cis-configured diastereoisomer in the reaction flask rather than thermodynamic equilibration. As such, this reaction represents an example of a crystallisation-induced diastereomeric transformation (CIDT) [104-108]. This is supported by the observation that 2m and 2m’, when exposed separately to simulated reaction conditions, epimerised at C5 to afford an identical 63:37 thermodynamic mixture of 2m/2m’(Scheme 7; Figure 3) [110].
![[1860-5397-8-64-i7]](/bjoc/content/inline/1860-5397-8-64-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Thermodynamically-driven epimerisation of 5-nitropiperidin-2-ones 2m and 2m’.
Scheme 7: Thermodynamically-driven epimerisation of 5-nitropiperidin-2-ones 2m and 2m’.
![[1860-5397-8-64-3]](/bjoc/content/figures/1860-5397-8-64-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Thermodynamically driven epimerisation of 5-nitropiperidin-2-ones 2m and 2m’; identical diastereomeric excess measured for both diastereomers after 48 h and 72 h.
Figure 3: Thermodynamically driven epimerisation of 5-nitropiperidin-2-ones 2m and 2m’; identical diastereome...
With all of the necessary variations to the nitro-Mannich/lactamisation cascade having been tested, optimised and scoped, we looked at the possibility of combining it with a catalytic asymmetric synthesis of a particular Michael adduct, so as to construct a one-pot enantio- and diastereoselective four-component coupling reaction (Scheme 8).
![[1860-5397-8-64-i8]](/bjoc/content/inline/1860-5397-8-64-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: One-pot three/four-component enantioselective Michael addition/nitro-Mannich/lactamisation cascade.
Scheme 8: One-pot three/four-component enantioselective Michael addition/nitro-Mannich/lactamisation cascade.
As described in our previous communication [61], the employment of bifunctional catalyst 9 [54,55] in a highly stereoselective two-stage one-pot cascade led to the formation of enantiomerically highly enriched spirocycle (+)-1a (Scheme 8). In a repeat of the process but with the intention of targeting a piperidin-2-one ring-containing polycyclic scaffold, cyclic imine 5a was added at the second stage. Tetracyclic spiro-lactam 2a was isolated in high enantiomeric purity (90% ee) in good chemical yield (62%, Scheme 8) [111,112].
For the products of the nitro-Mannich/lactamisation cascade to be of use in alkaloid natural-product synthesis (or even simple stereoselective piperidine synthesis), controlled, reductive manipulation of both the nitro group and the lactam carbonyl were required. Although Nef-type oxidation followed by exhaustive reduction of the resulting carbonyl group was considered, Ono’s radical procedure [113-116] was initially investigated. With some modification and optimisation, this was found to be compatible with the piperidin-2-one scaffold. Thus treatment of 2a and 2c with tributyltin hydride and AIBN in toluene under reflux smoothly afforded the protodenitrated products 10c and 10d in good yield (average 76% yield, Scheme 9). Other examples of successful nitro-group removal were also achieved when substrates 1i and 1j, lacking additional rings but bearing sensitive moieties (triple bond and furan moiety), were exposed to identical reaction conditions. The piperidin-2-ones 10a and 10b were obtained in 53% and 84%, respectively. The reduction of both piperidin-2-one and pyrrolidin-2-one heterocycles to piperidine or pyrrolidine rings by using a range of reagents is well-documented in the literature [117,118]. However, we believed that a controlled, chemoselective reduction would offer more options in any synthesis, and thus several commercially available reducing agents were screened in order to achieve selective reduction of only one lactam carbonyl. A notable find was that, by short exposure of denitrated heterocycle 10a–c to LiAlH4 in THF followed by quenching and treatment with HCOOH, spirocycles 11a–c were obtained in good yields. The chemoselectivity of the reduction was unambiguously confirmed by single-crystal X-ray diffraction studies of 11c. Furthermore, the use of an excess of DIBAL at room temperature smoothly afforded the diamines 12a and 12b (Scheme 10).
![[1860-5397-8-64-i9]](/bjoc/content/inline/1860-5397-8-64-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Protodenitration of 5-nitropiperidin-2-ones.
Scheme 9: Protodenitration of 5-nitropiperidin-2-ones.
![[1860-5397-8-64-i10]](/bjoc/content/inline/1860-5397-8-64-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Various reductions of denitrated heterocycles.
Scheme 10: Various reductions of denitrated heterocycles.
Conclusion
In summary, a versatile nitro-Mannich/lactamisation cascade for the direct synthesis of heavily decorated 5-nitropiperidin-2-ones and related heterocycles has been developed. A highly enantioenriched substituted 5-nitropiperidin-2-one was synthesised in a four-component one-pot cascade combining an enantioselective Michael addition with the diastereoselective nitro-Mannich/lactamisation cascade. Protodenitration and chemoselective reductive manipulation of the heterocycles could be used to install contiguous and fully substituted stereocentres in the synthesis of architecturally complex multicyclic alkaloid structures. The first applications of the developed methodology were disclosed recently as the total syntheses of paroxetine [60] and nakadomarin A [61-65] were successfully finished by employing the strategy as a fundamental synthetic tool. Further development is ongoing in our laboratory and the results will be disclosed in due course.
Supporting Information
| Supporting Information File 1: General experimental, copies of 1H and 13C NMR spectra for all new compounds (1a–j, 2a, 2a’’, 2m, 2m’, 6b–e, 10a–d, 11a–c, 12a,b). | ||
| Format: PDF | Size: 5.5 MB | Download |
| Supporting Information File 2: X-ray crystal structure of compound 2m’. | ||
| Format: CIF | Size: 23.2 KB | Download |
| Supporting Information File 3: X-ray crystal structures of compounds 1g, 2, 2a’’, 6e and 11c. | ||
| Format: CIF | Size: 97.7 KB | Download |
Acknowledgements
We gratefully acknowledge Merck, Sharp and Dohme (Hoddesdon, U.K.) for a studentship (to D.M.C.); the EPSRC for a studentship (to D.M.C), a postdoctoral fellowship (to P.J.), and a Leadership Fellowship (to D.J.D.). We thank Dr Edward Cleator of MSD for useful discussion, Andrew Kyle and Katherine Bogle for X-ray structure determination and the Oxford Chemical Crystallography Service for use of the instrumentation.
References
-
de Oliveira, J. H. H. L.; Nascimento, A. M.; Kossuga, M. H.; Cavalcanti, B. C.; Pessoa, C. O.; Moraes, M. O.; Macedo, M. L.; Ferreira, A. G.; Hajdu, E.; Pinheiro, U. S.; Berlinck, R. G. S. J. Nat. Prod. 2007, 70, 538–543. doi:10.1021/np060450q
Return to citation in text: [1] -
Magnier, E.; Langlois, Y. Tetrahedron 1998, 54, 6201–6258. doi:10.1016/S0040-4020(98)00357-3
See for a review on the isolation and the biological properties of manzamine A.
Return to citation in text: [1] -
Winkler, J. D.; Axten, J. M. J. Am. Chem. Soc. 1998, 120, 6425–6426. doi:10.1021/ja981303k
Return to citation in text: [1] -
Humphrey, J. M.; Liao, Y.; Ali, A.; Rein, T.; Wong, Y.-L.; Chen, H.-J.; Courtney, A. K.; Martin, S. F. J. Am. Chem. Soc. 2002, 124, 8584–8592. doi:10.1021/ja0202964
Return to citation in text: [1] -
Toma, T.; Kita, Y.; Fukuyama, T. J. Am. Chem. Soc. 2010, 132, 10233–10235. doi:10.1021/ja103721s
Return to citation in text: [1] -
Yamada, M.; Takahashi, Y.; Kubota, T.; Fromont, J.; Ishiyama, A.; Otoguro, K.; Yamada, H.; Ōmura, S.; Kobayashi, J.-i. Tetrahedron 2009, 65, 2313–2317. doi:10.1016/j.tet.2009.01.032
Return to citation in text: [1] -
Chen, F.-E.; Huang, J. Chem. Rev. 2005, 105, 4671–4706. doi:10.1021/cr050521a
Return to citation in text: [1] -
Woodward, R. B.; Bader, F. E.; Bickel, H.; Frey, A. J.; Kierstead, R. W. Tetrahedron 1958, 2, 1–57. doi:10.1016/0040-4020(58)88022-9
Return to citation in text: [1] -
Barnes, R. D.; Wodd-Kaczmar, M. W.; Curzons, A. D.; Lynch, I.; Richardson, J. E.; Buxton, P. C. Anti-depressant crystalline paroxetine hydrochloride hemihydrate. U.S. Patent 4,721,723, Oct 23, 1986.
Return to citation in text: [1] -
Yu, M. S.; Lantos, I.; Peng, Z.-Q.; Yu, J.; Cacchio, T. Tetrahedron Lett. 2000, 41, 5647–5651. doi:10.1016/S0040-4039(00)00942-4
Return to citation in text: [1] -
Furkert, D. P.; Husbands, S. M. Org. Lett. 2007, 9, 3769–3771. doi:10.1021/ol0713988
Return to citation in text: [1] -
Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957–5978. doi:10.1016/S0040-4020(02)00584-7
Return to citation in text: [1] -
Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193
Return to citation in text: [1] -
Bailey, P. D.; Millwood, P. A.; Smith, P. D. Chem. Commun. 1998, 633–640. doi:10.1039/a709071d
Return to citation in text: [1] -
O'Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d
Return to citation in text: [1] -
Buffat, M. G. P. Tetrahedron 2004, 60, 1701–1729. doi:10.1016/j.tet.2003.11.043
Return to citation in text: [1] -
Weintraub, P. M.; Sabol, J. S.; Kane, J. M.; Borcherding, D. R. Tetrahedron 2003, 59, 2953–2989. doi:10.1016/S0040-4020(03)00295-3
Return to citation in text: [1] -
Husson, H.-P.; Royer, J. Chem. Soc. Rev. 1999, 28, 383–394. doi:10.1039/a900153k
Return to citation in text: [1] -
Escolano, C.; Amat, M.; Bosch, J. Chem.–Eur. J. 2006, 12, 8198–8207. doi:10.1002/chem.200600813
Return to citation in text: [1] -
Comins, D. L.; Fulp, A. B. Tetrahedron Lett. 2001, 42, 6839–6841. doi:10.1016/S0040-4039(01)01432-0
Return to citation in text: [1] -
Comins, D. L.; Brooks, C. A.; Ingalls, C. L. J. Org. Chem. 2001, 66, 2181–2182. doi:10.1021/jo001609l
Return to citation in text: [1] -
Comins, D. L.; Libby, A. H.; Al-awar, R. S.; Foti, C. J. J. Org. Chem. 1999, 64, 2184–2185. doi:10.1021/jo990192k
Return to citation in text: [1] -
Comins, D. L.; Kuethe, J. T.; Miller, T. M.; Février, F. C.; Brooks, C. A. J. Org. Chem. 2005, 70, 5221–5234. doi:10.1021/jo050559n
Return to citation in text: [1] -
Kuethe, J. T.; Comins, D. L. Org. Lett. 2000, 2, 855–857. doi:10.1021/ol0056271
Return to citation in text: [1] -
Comins, D. L.; Zhang, Y.-m.; Joseph, S. P. Org. Lett. 1999, 1, 657–659. doi:10.1021/ol990738p
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E.; Wrobleski, A. D. J. Am. Chem. Soc. 2005, 127, 10186–10187. doi:10.1021/ja0533895
Return to citation in text: [1] -
Lin, N.-H.; Overman, L. E.; Rabinowitz, M. H.; Robinson, L. A.; Sharp, M. J.; Zablocki, J. J. Am. Chem. Soc. 1996, 118, 9062–9072. doi:10.1021/ja961641q
Return to citation in text: [1] -
Castro, P.; Overman, L. E.; Zhang, X.; Mariano, P. S. Tetrahedron Lett. 1993, 34, 5243–5246. doi:10.1016/S0040-4039(00)73963-3
Return to citation in text: [1] -
Hong, C. Y.; Kado, N.; Overman, L. E. J. Am. Chem. Soc. 1993, 115, 11028–11029. doi:10.1021/ja00076a086
Return to citation in text: [1] -
Gedig, T.; Dettner, K.; Seifert, K. Tetrahedron 2007, 63, 2670–2674. doi:10.1016/j.tet.2007.01.024
Return to citation in text: [1] -
Trost, B. M.; Cramer, N.; Bernsmann, H. J. Am. Chem. Soc. 2007, 129, 3086–3087. doi:10.1021/ja070142u
Return to citation in text: [1] -
Islam, I.; Bryant, J.; May, K.; Mohan, R.; Yuan, S.; Kent, L.; Morser, J.; Zhao, L.; Vergona, R.; White, K.; Adler, M.; Whitlow, M.; Buckman, B. O. Bioorg. Med. Chem. Lett. 2007, 17, 1349–1354. doi:10.1016/j.bmcl.2006.11.078
Return to citation in text: [1] -
Pandey, S. K.; Kumar, P. Tetrahedron Lett. 2005, 46, 4091–4093. doi:10.1016/j.tetlet.2005.04.013
Return to citation in text: [1] -
Takahata, H.; Saito, Y.; Ichinose, M. Org. Biomol. Chem. 2006, 4, 1587–1595. doi:10.1039/B601489E
Return to citation in text: [1] -
Kim, S.-G.; Lee, S. H.; Park, T.-H. Tetrahedron Lett. 2007, 48, 5023–5026. doi:10.1016/j.tetlet.2007.05.100
Return to citation in text: [1] -
Breuning, M.; Steiner, M. Synthesis 2006, 1386–1389. doi:10.1055/s-2006-926419
Return to citation in text: [1] -
Itoh, T.; Nishimura, K.; Nagata, K.; Yokoya, M. Synlett 2006, 2207–2210. doi:10.1055/s-2006-948203
Return to citation in text: [1] -
Gebauer, J.; Blechert, S. Synlett 2005, 2826–2828. doi:10.1055/s-2005-918942
Return to citation in text: [1] -
Enders, D.; Thiebes, C. Pure Appl. Chem. 2001, 73, 573–578. doi:10.1351/pac200173030573
Return to citation in text: [1] -
Davis, F. A.; Santhanaraman, M. J. Org. Chem. 2006, 71, 4222–4226. doi:10.1021/jo060371j
Return to citation in text: [1] -
Ciblat, S.; Besse, P.; Papastergiou, V.; Veschambre, H.; Canet, J.-L.; Troin, Y. Tetrahedron: Asymmetry 2000, 11, 2221–2229. doi:10.1016/S0957-4166(00)00162-2
Return to citation in text: [1] -
Jo, E.; Na, Y.; Chang, S. Tetrahedron Lett. 1999, 40, 5581–5582. doi:10.1016/S0040-4039(99)01081-3
Return to citation in text: [1] -
Schaudt, M.; Blechert, S. J. Org. Chem. 2003, 68, 2913–2920. doi:10.1021/jo026803h
Return to citation in text: [1] -
Nicolau, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. doi:10.1002/anie.200500369
Return to citation in text: [1] -
Takahata, H.; Banba, Y.; Ouchi, H.; Nemoto, H.; Kato, A.; Adachi, I. J. Org. Chem. 2003, 68, 3603–3607. doi:10.1021/jo034137u
Return to citation in text: [1] -
Kim, I. S.; Oh, J. S.; Zee, O. P.; Jung, Y. H. Tetrahedron 2007, 63, 2622–2633. doi:10.1016/j.tet.2007.01.028
Return to citation in text: [1] -
Felpin, F.-X.; Girard, S.; Vo-Thanh, G.; Robins, R. J.; Villiéras, J.; Lebreton, J. J. Org. Chem. 2001, 66, 6305–6312. doi:10.1021/jo010386b
Return to citation in text: [1] -
Mulzer, J.; Öhler, E. Top. Organomet. Chem. 2004, 13, 269–366. doi:10.1007/b98768
Return to citation in text: [1] -
Kim, S.; Lee, Y. M.; Lee, J.; Lee, T.; Fu, Y.; Song, Y.; Cho, J.; Kim, D. J. Org. Chem. 2007, 72, 4886–4891. doi:10.1021/jo070668x
Return to citation in text: [1] -
Goodenough, K. M.; Moran, W. J.; Raubo, P.; Harrity, J. P. A. J. Org. Chem. 2005, 70, 207–213. doi:10.1021/jo048455k
Return to citation in text: [1] -
Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899–4902. doi:10.1021/ol0619359
Return to citation in text: [1] -
Harrity, J. P. A.; Provoost, O. Org. Biomol. Chem. 2005, 3, 1349–1358. doi:10.1039/b502349c
Return to citation in text: [1] -
Itoh, J.; Fuchibe, K.; Akiyama, T. Angew. Chem., Int. Ed. 2006, 45, 4796–4798. doi:10.1002/anie.200601345
Return to citation in text: [1] -
Hynes, P. S.; Stranges, D.; Stupple, P. A.; Guarna, A.; Dixon, D. J. Org. Lett. 2007, 9, 2107–2110. doi:10.1021/ol070532l
Return to citation in text: [1] [2] -
Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j
Return to citation in text: [1] [2] -
Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672–12673. doi:10.1021/ja036972z
Return to citation in text: [1] -
Li, H.; Wang, Y.; Tang, L.; Deng, L. J. Am. Chem. Soc. 2004, 126, 9906–9907. doi:10.1021/ja047281l
Return to citation in text: [1] -
McCooey, S. H.; Connon, S. J. Angew. Chem., Int. Ed. 2005, 44, 6367–6370. doi:10.1002/anie.200501721
Return to citation in text: [1] -
Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/b719249e
Return to citation in text: [1] -
Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389–1391. doi:10.1021/ol800108u
Return to citation in text: [1] [2] -
Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s
Return to citation in text: [1] [2] -
Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h
Return to citation in text: [1] [2] -
Jakubec, P.; Kyle, A. F.; Calleja, J.; Dixon, D. J. Tetrahedron Lett. 2011, 52, 6094–6097. doi:10.1016/j.tetlet.2011.09.016
Return to citation in text: [1] [2] -
Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563
Return to citation in text: [1] [2] -
Mühlstädt, M.; Schulze, B. J. Prakt. Chem. 1975, 317, 919–925. doi:10.1002/prac.19753170606
Return to citation in text: [1] -
Bhagwatheeswaran, H.; Gaur, S. P.; Jain, P. C. Synthesis 1976, 615–616. doi:10.1055/s-1976-24142
Return to citation in text: [1] -
Desai, M. C.; Thadeio, P. F.; Lefkowitz, S. L. Tetrahedron Lett. 1993, 34, 5831–5834. doi:10.1016/S0040-4039(00)73791-9
Return to citation in text: [1] -
Desai, M. C.; Lefkowitz, S. L. Bioorg. Med. Chem. Lett. 1993, 3, 2083–2086. doi:10.1016/S0960-894X(01)81021-0
Return to citation in text: [1] -
Nara, S.; Tanaka, R.; Eishima, J.; Hara, M.; Takahashi, Y.; Otaki, S.; Foglesong, R. J.; Hughes, P. F.; Turkington, S.; Kanda, Y. J. Med. Chem. 2003, 46, 2467–2473. doi:10.1021/jm020522k
Return to citation in text: [1] -
Tanaka, R.; Rubio, A.; Harn, N. K.; Gernert, D.; Grese, T. A.; Eishima, J.; Hara, M.; Yoda, N.; Ohashi, R.; Kuwabara, T.; Soga, S.; Akinaga, S.; Nara, S.; Kanda, Y. Bioorg. Med. Chem. 2007, 15, 1363–1382. doi:10.1016/j.bmc.2006.11.007
Return to citation in text: [1] -
Pei, Z.; Li, X.; von Geldern, T. W.; Longenecker, K.; Pireh, D.; Stewart, K. D.; Backes, B. J.; Lai, C.; Lubben, T. H.; Ballaron, S. J.; Beno, D. W. A.; Kempf-Grote, A. J.; Sham, H. L.; Trevillyan, J. M. J. Med. Chem. 2007, 50, 1983–1987. doi:10.1021/jm061436d
Return to citation in text: [1] -
Xu, F.; Corley, E.; Zacuto, M.; Conlon, D. A.; Pipik, B.; Humphrey, G.; Murry, J.; Tschaen, D. J. Org. Chem. 2010, 75, 1343–1353. doi:10.1021/jo902573q
Return to citation in text: [1] -
Xu, F.; Corley, E.; Murry, J. A.; Tschaen, D. M. Org. Lett. 2007, 9, 2669–2672. doi:10.1021/ol070909n
Return to citation in text: [1] -
Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2009, 11, 4512–4515. doi:10.1021/ol901640v
Return to citation in text: [1] -
Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2011, 13, 6406–6409. doi:10.1021/ol202710g
Return to citation in text: [1] -
Barber, D. M.; Sanganee, H.; Dixon, D. J. Chem. Commun. 2011, 47, 4379–4381. doi:10.1039/c1cc10751h
Return to citation in text: [1] -
Rychnovsky, S. D.; Beauchamp, T.; Vaidyanathan, R.; Kwan, T. J. Org. Chem. 1998, 63, 6363–6374. doi:10.1021/jo9808831
Return to citation in text: [1] [2] -
Westermann, B. Angew. Chem., Int. Ed. 2003, 42, 151–153. doi:10.1002/anie.200390071
Return to citation in text: [1] [2] -
Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626–2704. doi:10.1021/cr100204f
Return to citation in text: [1] [2] -
Humphrey, J. M.; Arnold, E. P.; Chappie, T. A.; Feltenberger, J. B.; Nagel, A.; Simon, W.; Suarez-Contreras, M.; Tom, N. J.; O’Neill, B. T. J. Org. Chem. 2009, 74, 4525–4536. doi:10.1021/jo9003184
Return to citation in text: [1] -
Imashiro, R.; Uehara, H.; Barbas, C. F., III. Org. Lett. 2010, 12, 5250–5253. doi:10.1021/ol102292a
Return to citation in text: [1] -
Wang, Y.; Yu, D.-F.; Liu, Y.-Z.; Wei, H.; Luo, Y.-C.; Dixon, D. J.; Xu, P.-F. Chem.–Eur. J. 2010, 16, 3922–3925. doi:10.1002/chem.201000059
Return to citation in text: [1] -
Only one diastereomer was observed by 1H NMR.
Return to citation in text: [1] -
Elliot, M. C.; Williams, E. Org. Biomol. Chem. 2003, 1, 3038–3047. doi:10.1039/b306159k
Return to citation in text: [1] -
Struve, C.; Christophersen, C. Heterocycles 2003, 60, 1907–1914. doi:10.3987/COM-03-9802
Return to citation in text: [1] -
Scully, F. E., Jr. J. Org. Chem. 1980, 45, 1515–1517. doi:10.1021/jo01296a036
Return to citation in text: [1] -
Davis, B. G.; Maughan, M. A. T.; Chapman, T. M.; Villard, R.; Courtney, S. Org. Lett. 2002, 4, 103–106. doi:10.1021/ol016970o
Return to citation in text: [1] -
Bertrand, M.; Poissonnet, G.; Théret-Bettiol, M.-H.; Gaspard, C.; Werner, G. H.; Pfeiffer, B.; Renard, P.; Léonce, S.; Dodd, R. H. Bioorg. Med. Chem. 2001, 9, 2155–2164. doi:10.1016/S0968-0896(01)00119-5
Return to citation in text: [1] -
Warrener, R. N.; Liu, L.; Russell, R. A. Tetrahedron 1998, 54, 7485–7496. doi:10.1016/S0040-4020(98)00378-0
Return to citation in text: [1] -
Caroon, J. M.; Clark, R. D.; Kluge, A. F.; Lee, C. H.; Strosberg, A. M. J. Med. Chem. 1983, 26, 1426–1433. doi:10.1021/jm00364a013
Return to citation in text: [1] -
Venkov, A. P.; Statkova-Abeghe, S. Synth. Commun. 1996, 26, 127–134. doi:10.1080/00397919608003871
Return to citation in text: [1] -
Grunewald, G. L.; Dahanukar, V. H.; Ching, P.; Criscione, K. R. J. Med. Chem. 1996, 39, 3539–3546. doi:10.1021/jm9508292
Return to citation in text: [1] -
Meyers, A. I.; Hutchings, R. H. Tetrahedron 1993, 49, 1807–1820. doi:10.1016/S0040-4020(01)80537-8
Return to citation in text: [1] -
García, E.; Lete, E.; Sotomayor, N. J. Org. Chem. 2006, 71, 6776–6784. doi:10.1021/jo060903w
Return to citation in text: [1] -
Itoh, T.; Miyazaki, M.; Fukuoka, H.; Nagata, K.; Ohsawa, A. Org. Lett. 2006, 8, 1295–1297. doi:10.1021/ol0530326
Return to citation in text: [1] -
Ma, J.; Yin, W.; Zhou, H.; Cook, J. M. Org. Lett. 2007, 9, 3491–3494. doi:10.1021/ol071220l
Return to citation in text: [1] -
Sheludko, Y.; Gerasimenko, I.; Kolshorn, H.; Stöckigt, J. J. Nat. Prod. 2002, 65, 1006–1010. doi:10.1021/np0200919
Return to citation in text: [1] -
Bailey, P. D.; Morgan, K. M. J. Chem. Soc., Perkin Trans. 1 2000, 3578–3583. doi:10.1039/B005695M
Return to citation in text: [1] -
Batista, C. V. F.; Schripsema, J.; Verpoorte, R.; Rech, S. B.; Henriques, A. T. Phytochemistry 1996, 41, 969–973. doi:10.1016/0031-9422(95)00666-4
Return to citation in text: [1] -
Kuehne, M. E.; Muth, R. S. J. Org. Chem. 1991, 56, 2701–2712. doi:10.1021/jo00008a025
Return to citation in text: [1] -
Martin, S. F.; Benage, B.; Hunter, J. E. J. Am. Chem. Soc. 1988, 110, 5925–5927. doi:10.1021/ja00225a068
Return to citation in text: [1] -
The relative stereochemistry was confirmed by the measurement of J-coupling constants and unambiguously determined by single-crystal X-ray analysis.
Return to citation in text: [1] -
Eliel, E. L.; Wilen, S. H.; Mander, L. N. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 1994.
See for the definition of crystallisation-induced transformations.
Return to citation in text: [1] [2] -
Yoshioka, R. Top. Curr. Chem. 2007, 269, 83–132. doi:10.1007/128_2006_094
Return to citation in text: [1] [2] -
Brands, K. M. J.; Davies, A. J. Chem. Rev. 2006, 106, 2711–2733. doi:10.1021/cr0406864
Return to citation in text: [1] [2] -
Anderson, N. G. Org. Process Res. Dev. 2005, 9, 800–813. doi:10.1021/op050119y
Return to citation in text: [1] [2] -
Ďuriš, A.; Wiesenganger, T.; Moravčíková, D.; Baran, P.; Kožíšek, J.; Daïch, A.; Berkeš, D. Org. Lett. 2011, 13, 1642–1645. doi:10.1021/ol2001057
Return to citation in text: [1] [2] -
Adams, H.; Anderson, J. C.; Peace, S.; Pennell, A. M. K. J. Org. Chem. 1998, 63, 9932–9934. doi:10.1021/jo981700d
Return to citation in text: [1] -
The epimerisation studies were performed in MeOH (c 0.015 M) to ensure the full solubility of all reagents and products.
Return to citation in text: [1] -
Absolute stereochemistry assigned by analogy with previous results of Michael addition to nitroolefin electrophiles catalyzed by 9; see reference [55]
Return to citation in text: [1] -
Dixon, D. J.; Ley, S. V.; Rodríguez, F. Angew. Chem., Int. Ed. 2001, 40, 4763–4765. doi:10.1002/1521-3773(20011217)40:24<4763::AID-ANIE4763>3.0.CO;2-D
See for a related multicomponent reaction.
Return to citation in text: [1] -
Ono, N.; Miyake, H.; Tamura, R.; Kaji, A. Tetrahedron Lett. 1981, 22, 1705–1708. doi:10.1016/S0040-4039(01)90417-4
See for seminal work.
Return to citation in text: [1] -
Ono, N.; Kaji, A. Synthesis 1986, 693–704. doi:10.1055/s-1986-31754
Return to citation in text: [1] -
Tormo, J.; Hays, D. S.; Fu, G. C. J. Org. Chem. 1998, 63, 5296–5297. doi:10.1021/jo980789k
Return to citation in text: [1] -
Shen, B.; Johnston, J. N. Org. Lett. 2008, 10, 4397–4400. doi:10.1021/ol801797h
Return to citation in text: [1] -
Meyers, A. I.; Downing, S. V.; Weiser, M. J. J. Org. Chem. 2001, 66, 1413–1419. doi:10.1021/jo001548r
Return to citation in text: [1] -
Hirosawa, C.; Wakasa, N.; Fuchikami, T. Tetrahedron Lett. 1996, 37, 6749–6752. doi:10.1016/S0040-4039(96)01458-X
Return to citation in text: [1]
| 61. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 86. | Struve, C.; Christophersen, C. Heterocycles 2003, 60, 1907–1914. doi:10.3987/COM-03-9802 |
| 87. | Scully, F. E., Jr. J. Org. Chem. 1980, 45, 1515–1517. doi:10.1021/jo01296a036 |
| 88. | Davis, B. G.; Maughan, M. A. T.; Chapman, T. M.; Villard, R.; Courtney, S. Org. Lett. 2002, 4, 103–106. doi:10.1021/ol016970o |
| 89. | Bertrand, M.; Poissonnet, G.; Théret-Bettiol, M.-H.; Gaspard, C.; Werner, G. H.; Pfeiffer, B.; Renard, P.; Léonce, S.; Dodd, R. H. Bioorg. Med. Chem. 2001, 9, 2155–2164. doi:10.1016/S0968-0896(01)00119-5 |
| 90. | Warrener, R. N.; Liu, L.; Russell, R. A. Tetrahedron 1998, 54, 7485–7496. doi:10.1016/S0040-4020(98)00378-0 |
| 91. | Caroon, J. M.; Clark, R. D.; Kluge, A. F.; Lee, C. H.; Strosberg, A. M. J. Med. Chem. 1983, 26, 1426–1433. doi:10.1021/jm00364a013 |
| 92. | Venkov, A. P.; Statkova-Abeghe, S. Synth. Commun. 1996, 26, 127–134. doi:10.1080/00397919608003871 |
| 93. | Grunewald, G. L.; Dahanukar, V. H.; Ching, P.; Criscione, K. R. J. Med. Chem. 1996, 39, 3539–3546. doi:10.1021/jm9508292 |
| 94. | Meyers, A. I.; Hutchings, R. H. Tetrahedron 1993, 49, 1807–1820. doi:10.1016/S0040-4020(01)80537-8 |
| 95. | García, E.; Lete, E.; Sotomayor, N. J. Org. Chem. 2006, 71, 6776–6784. doi:10.1021/jo060903w |
| 96. | Itoh, T.; Miyazaki, M.; Fukuoka, H.; Nagata, K.; Ohsawa, A. Org. Lett. 2006, 8, 1295–1297. doi:10.1021/ol0530326 |
| 97. | Ma, J.; Yin, W.; Zhou, H.; Cook, J. M. Org. Lett. 2007, 9, 3491–3494. doi:10.1021/ol071220l |
| 98. | Sheludko, Y.; Gerasimenko, I.; Kolshorn, H.; Stöckigt, J. J. Nat. Prod. 2002, 65, 1006–1010. doi:10.1021/np0200919 |
| 99. | Bailey, P. D.; Morgan, K. M. J. Chem. Soc., Perkin Trans. 1 2000, 3578–3583. doi:10.1039/B005695M |
| 100. | Batista, C. V. F.; Schripsema, J.; Verpoorte, R.; Rech, S. B.; Henriques, A. T. Phytochemistry 1996, 41, 969–973. doi:10.1016/0031-9422(95)00666-4 |
| 101. | Kuehne, M. E.; Muth, R. S. J. Org. Chem. 1991, 56, 2701–2712. doi:10.1021/jo00008a025 |
| 102. | Martin, S. F.; Benage, B.; Hunter, J. E. J. Am. Chem. Soc. 1988, 110, 5925–5927. doi:10.1021/ja00225a068 |
| 61. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 1. | de Oliveira, J. H. H. L.; Nascimento, A. M.; Kossuga, M. H.; Cavalcanti, B. C.; Pessoa, C. O.; Moraes, M. O.; Macedo, M. L.; Ferreira, A. G.; Hajdu, E.; Pinheiro, U. S.; Berlinck, R. G. S. J. Nat. Prod. 2007, 70, 538–543. doi:10.1021/np060450q |
| 11. | Furkert, D. P.; Husbands, S. M. Org. Lett. 2007, 9, 3769–3771. doi:10.1021/ol0713988 |
| 60. | Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389–1391. doi:10.1021/ol800108u |
| 61. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 9. | Barnes, R. D.; Wodd-Kaczmar, M. W.; Curzons, A. D.; Lynch, I.; Richardson, J. E.; Buxton, P. C. Anti-depressant crystalline paroxetine hydrochloride hemihydrate. U.S. Patent 4,721,723, Oct 23, 1986. |
| 10. | Yu, M. S.; Lantos, I.; Peng, Z.-Q.; Yu, J.; Cacchio, T. Tetrahedron Lett. 2000, 41, 5647–5651. doi:10.1016/S0040-4039(00)00942-4 |
| 61. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 54. | Hynes, P. S.; Stranges, D.; Stupple, P. A.; Guarna, A.; Dixon, D. J. Org. Lett. 2007, 9, 2107–2110. doi:10.1021/ol070532l |
| 55. | Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j |
| 7. | Chen, F.-E.; Huang, J. Chem. Rev. 2005, 105, 4671–4706. doi:10.1021/cr050521a |
| 8. | Woodward, R. B.; Bader, F. E.; Bickel, H.; Frey, A. J.; Kierstead, R. W. Tetrahedron 1958, 2, 1–57. doi:10.1016/0040-4020(58)88022-9 |
| 49. | Kim, S.; Lee, Y. M.; Lee, J.; Lee, T.; Fu, Y.; Song, Y.; Cho, J.; Kim, D. J. Org. Chem. 2007, 72, 4886–4891. doi:10.1021/jo070668x |
| 50. | Goodenough, K. M.; Moran, W. J.; Raubo, P.; Harrity, J. P. A. J. Org. Chem. 2005, 70, 207–213. doi:10.1021/jo048455k |
| 51. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899–4902. doi:10.1021/ol0619359 |
| 52. | Harrity, J. P. A.; Provoost, O. Org. Biomol. Chem. 2005, 3, 1349–1358. doi:10.1039/b502349c |
| 53. | Itoh, J.; Fuchibe, K.; Akiyama, T. Angew. Chem., Int. Ed. 2006, 45, 4796–4798. doi:10.1002/anie.200601345 |
| 104. |
Eliel, E. L.; Wilen, S. H.; Mander, L. N. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 1994.
See for the definition of crystallisation-induced transformations. |
| 105. | Yoshioka, R. Top. Curr. Chem. 2007, 269, 83–132. doi:10.1007/128_2006_094 |
| 106. | Brands, K. M. J.; Davies, A. J. Chem. Rev. 2006, 106, 2711–2733. doi:10.1021/cr0406864 |
| 107. | Anderson, N. G. Org. Process Res. Dev. 2005, 9, 800–813. doi:10.1021/op050119y |
| 108. | Ďuriš, A.; Wiesenganger, T.; Moravčíková, D.; Baran, P.; Kožíšek, J.; Daïch, A.; Berkeš, D. Org. Lett. 2011, 13, 1642–1645. doi:10.1021/ol2001057 |
| 2. |
Magnier, E.; Langlois, Y. Tetrahedron 1998, 54, 6201–6258. doi:10.1016/S0040-4020(98)00357-3
See for a review on the isolation and the biological properties of manzamine A. |
| 3. | Winkler, J. D.; Axten, J. M. J. Am. Chem. Soc. 1998, 120, 6425–6426. doi:10.1021/ja981303k |
| 4. | Humphrey, J. M.; Liao, Y.; Ali, A.; Rein, T.; Wong, Y.-L.; Chen, H.-J.; Courtney, A. K.; Martin, S. F. J. Am. Chem. Soc. 2002, 124, 8584–8592. doi:10.1021/ja0202964 |
| 5. | Toma, T.; Kita, Y.; Fukuyama, T. J. Am. Chem. Soc. 2010, 132, 10233–10235. doi:10.1021/ja103721s |
| 6. | Yamada, M.; Takahashi, Y.; Kubota, T.; Fromont, J.; Ishiyama, A.; Otoguro, K.; Yamada, H.; Ōmura, S.; Kobayashi, J.-i. Tetrahedron 2009, 65, 2313–2317. doi:10.1016/j.tet.2009.01.032 |
| 54. | Hynes, P. S.; Stranges, D.; Stupple, P. A.; Guarna, A.; Dixon, D. J. Org. Lett. 2007, 9, 2107–2110. doi:10.1021/ol070532l |
| 55. | Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j |
| 56. | Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672–12673. doi:10.1021/ja036972z |
| 57. | Li, H.; Wang, Y.; Tang, L.; Deng, L. J. Am. Chem. Soc. 2004, 126, 9906–9907. doi:10.1021/ja047281l |
| 58. | McCooey, S. H.; Connon, S. J. Angew. Chem., Int. Ed. 2005, 44, 6367–6370. doi:10.1002/anie.200501721 |
| 59. | Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/b719249e |
| 110. | The epimerisation studies were performed in MeOH (c 0.015 M) to ensure the full solubility of all reagents and products. |
| 30. | Gedig, T.; Dettner, K.; Seifert, K. Tetrahedron 2007, 63, 2670–2674. doi:10.1016/j.tet.2007.01.024 |
| 31. | Trost, B. M.; Cramer, N.; Bernsmann, H. J. Am. Chem. Soc. 2007, 129, 3086–3087. doi:10.1021/ja070142u |
| 32. | Islam, I.; Bryant, J.; May, K.; Mohan, R.; Yuan, S.; Kent, L.; Morser, J.; Zhao, L.; Vergona, R.; White, K.; Adler, M.; Whitlow, M.; Buckman, B. O. Bioorg. Med. Chem. Lett. 2007, 17, 1349–1354. doi:10.1016/j.bmcl.2006.11.078 |
| 38. | Gebauer, J.; Blechert, S. Synlett 2005, 2826–2828. doi:10.1055/s-2005-918942 |
| 39. | Enders, D.; Thiebes, C. Pure Appl. Chem. 2001, 73, 573–578. doi:10.1351/pac200173030573 |
| 40. | Davis, F. A.; Santhanaraman, M. J. Org. Chem. 2006, 71, 4222–4226. doi:10.1021/jo060371j |
| 41. | Ciblat, S.; Besse, P.; Papastergiou, V.; Veschambre, H.; Canet, J.-L.; Troin, Y. Tetrahedron: Asymmetry 2000, 11, 2221–2229. doi:10.1016/S0957-4166(00)00162-2 |
| 104. |
Eliel, E. L.; Wilen, S. H.; Mander, L. N. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 1994.
See for the definition of crystallisation-induced transformations. |
| 105. | Yoshioka, R. Top. Curr. Chem. 2007, 269, 83–132. doi:10.1007/128_2006_094 |
| 106. | Brands, K. M. J.; Davies, A. J. Chem. Rev. 2006, 106, 2711–2733. doi:10.1021/cr0406864 |
| 107. | Anderson, N. G. Org. Process Res. Dev. 2005, 9, 800–813. doi:10.1021/op050119y |
| 108. | Ďuriš, A.; Wiesenganger, T.; Moravčíková, D.; Baran, P.; Kožíšek, J.; Daïch, A.; Berkeš, D. Org. Lett. 2011, 13, 1642–1645. doi:10.1021/ol2001057 |
| 26. | Dounay, A. B.; Overman, L. E.; Wrobleski, A. D. J. Am. Chem. Soc. 2005, 127, 10186–10187. doi:10.1021/ja0533895 |
| 27. | Lin, N.-H.; Overman, L. E.; Rabinowitz, M. H.; Robinson, L. A.; Sharp, M. J.; Zablocki, J. J. Am. Chem. Soc. 1996, 118, 9062–9072. doi:10.1021/ja961641q |
| 28. | Castro, P.; Overman, L. E.; Zhang, X.; Mariano, P. S. Tetrahedron Lett. 1993, 34, 5243–5246. doi:10.1016/S0040-4039(00)73963-3 |
| 29. | Hong, C. Y.; Kado, N.; Overman, L. E. J. Am. Chem. Soc. 1993, 115, 11028–11029. doi:10.1021/ja00076a086 |
| 42. | Jo, E.; Na, Y.; Chang, S. Tetrahedron Lett. 1999, 40, 5581–5582. doi:10.1016/S0040-4039(99)01081-3 |
| 43. | Schaudt, M.; Blechert, S. J. Org. Chem. 2003, 68, 2913–2920. doi:10.1021/jo026803h |
| 44. | Nicolau, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. doi:10.1002/anie.200500369 |
| 45. | Takahata, H.; Banba, Y.; Ouchi, H.; Nemoto, H.; Kato, A.; Adachi, I. J. Org. Chem. 2003, 68, 3603–3607. doi:10.1021/jo034137u |
| 46. | Kim, I. S.; Oh, J. S.; Zee, O. P.; Jung, Y. H. Tetrahedron 2007, 63, 2622–2633. doi:10.1016/j.tet.2007.01.028 |
| 47. | Felpin, F.-X.; Girard, S.; Vo-Thanh, G.; Robins, R. J.; Villiéras, J.; Lebreton, J. J. Org. Chem. 2001, 66, 6305–6312. doi:10.1021/jo010386b |
| 48. | Mulzer, J.; Öhler, E. Top. Organomet. Chem. 2004, 13, 269–366. doi:10.1007/b98768 |
| 109. | Adams, H.; Anderson, J. C.; Peace, S.; Pennell, A. M. K. J. Org. Chem. 1998, 63, 9932–9934. doi:10.1021/jo981700d |
| 20. | Comins, D. L.; Fulp, A. B. Tetrahedron Lett. 2001, 42, 6839–6841. doi:10.1016/S0040-4039(01)01432-0 |
| 21. | Comins, D. L.; Brooks, C. A.; Ingalls, C. L. J. Org. Chem. 2001, 66, 2181–2182. doi:10.1021/jo001609l |
| 22. | Comins, D. L.; Libby, A. H.; Al-awar, R. S.; Foti, C. J. J. Org. Chem. 1999, 64, 2184–2185. doi:10.1021/jo990192k |
| 23. | Comins, D. L.; Kuethe, J. T.; Miller, T. M.; Février, F. C.; Brooks, C. A. J. Org. Chem. 2005, 70, 5221–5234. doi:10.1021/jo050559n |
| 24. | Kuethe, J. T.; Comins, D. L. Org. Lett. 2000, 2, 855–857. doi:10.1021/ol0056271 |
| 25. | Comins, D. L.; Zhang, Y.-m.; Joseph, S. P. Org. Lett. 1999, 1, 657–659. doi:10.1021/ol990738p |
| 103. | The relative stereochemistry was confirmed by the measurement of J-coupling constants and unambiguously determined by single-crystal X-ray analysis. |
| 12. | Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957–5978. doi:10.1016/S0040-4020(02)00584-7 |
| 13. | Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193 |
| 14. | Bailey, P. D.; Millwood, P. A.; Smith, P. D. Chem. Commun. 1998, 633–640. doi:10.1039/a709071d |
| 15. | O'Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d |
| 16. | Buffat, M. G. P. Tetrahedron 2004, 60, 1701–1729. doi:10.1016/j.tet.2003.11.043 |
| 17. | Weintraub, P. M.; Sabol, J. S.; Kane, J. M.; Borcherding, D. R. Tetrahedron 2003, 59, 2953–2989. doi:10.1016/S0040-4020(03)00295-3 |
| 18. | Husson, H.-P.; Royer, J. Chem. Soc. Rev. 1999, 28, 383–394. doi:10.1039/a900153k |
| 19. | Escolano, C.; Amat, M.; Bosch, J. Chem.–Eur. J. 2006, 12, 8198–8207. doi:10.1002/chem.200600813 |
| 33. | Pandey, S. K.; Kumar, P. Tetrahedron Lett. 2005, 46, 4091–4093. doi:10.1016/j.tetlet.2005.04.013 |
| 34. | Takahata, H.; Saito, Y.; Ichinose, M. Org. Biomol. Chem. 2006, 4, 1587–1595. doi:10.1039/B601489E |
| 35. | Kim, S.-G.; Lee, S. H.; Park, T.-H. Tetrahedron Lett. 2007, 48, 5023–5026. doi:10.1016/j.tetlet.2007.05.100 |
| 36. | Breuning, M.; Steiner, M. Synthesis 2006, 1386–1389. doi:10.1055/s-2006-926419 |
| 37. | Itoh, T.; Nishimura, K.; Nagata, K.; Yokoya, M. Synlett 2006, 2207–2210. doi:10.1055/s-2006-948203 |
| 78. | Rychnovsky, S. D.; Beauchamp, T.; Vaidyanathan, R.; Kwan, T. J. Org. Chem. 1998, 63, 6363–6374. doi:10.1021/jo9808831 |
| 79. | Westermann, B. Angew. Chem., Int. Ed. 2003, 42, 151–153. doi:10.1002/anie.200390071 |
| 80. | Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626–2704. doi:10.1021/cr100204f |
| 68. | Desai, M. C.; Thadeio, P. F.; Lefkowitz, S. L. Tetrahedron Lett. 1993, 34, 5831–5834. doi:10.1016/S0040-4039(00)73791-9 |
| 69. | Desai, M. C.; Lefkowitz, S. L. Bioorg. Med. Chem. Lett. 1993, 3, 2083–2086. doi:10.1016/S0960-894X(01)81021-0 |
| 62. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s |
| 63. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h |
| 64. | Jakubec, P.; Kyle, A. F.; Calleja, J.; Dixon, D. J. Tetrahedron Lett. 2011, 52, 6094–6097. doi:10.1016/j.tetlet.2011.09.016 |
| 65. | Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563 |
| 111. | Absolute stereochemistry assigned by analogy with previous results of Michael addition to nitroolefin electrophiles catalyzed by 9; see reference [55] |
| 112. |
Dixon, D. J.; Ley, S. V.; Rodríguez, F. Angew. Chem., Int. Ed. 2001, 40, 4763–4765. doi:10.1002/1521-3773(20011217)40:24<4763::AID-ANIE4763>3.0.CO;2-D
See for a related multicomponent reaction. |
| 66. | Mühlstädt, M.; Schulze, B. J. Prakt. Chem. 1975, 317, 919–925. doi:10.1002/prac.19753170606 |
| 67. | Bhagwatheeswaran, H.; Gaur, S. P.; Jain, P. C. Synthesis 1976, 615–616. doi:10.1055/s-1976-24142 |
| 113. |
Ono, N.; Miyake, H.; Tamura, R.; Kaji, A. Tetrahedron Lett. 1981, 22, 1705–1708. doi:10.1016/S0040-4039(01)90417-4
See for seminal work. |
| 114. | Ono, N.; Kaji, A. Synthesis 1986, 693–704. doi:10.1055/s-1986-31754 |
| 115. | Tormo, J.; Hays, D. S.; Fu, G. C. J. Org. Chem. 1998, 63, 5296–5297. doi:10.1021/jo980789k |
| 116. | Shen, B.; Johnston, J. N. Org. Lett. 2008, 10, 4397–4400. doi:10.1021/ol801797h |
| 117. | Meyers, A. I.; Downing, S. V.; Weiser, M. J. J. Org. Chem. 2001, 66, 1413–1419. doi:10.1021/jo001548r |
| 118. | Hirosawa, C.; Wakasa, N.; Fuchikami, T. Tetrahedron Lett. 1996, 37, 6749–6752. doi:10.1016/S0040-4039(96)01458-X |
| 61. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 85. | Elliot, M. C.; Williams, E. Org. Biomol. Chem. 2003, 1, 3038–3047. doi:10.1039/b306159k |
| 81. | Humphrey, J. M.; Arnold, E. P.; Chappie, T. A.; Feltenberger, J. B.; Nagel, A.; Simon, W.; Suarez-Contreras, M.; Tom, N. J.; O’Neill, B. T. J. Org. Chem. 2009, 74, 4525–4536. doi:10.1021/jo9003184 |
| 82. | Imashiro, R.; Uehara, H.; Barbas, C. F., III. Org. Lett. 2010, 12, 5250–5253. doi:10.1021/ol102292a |
| 83. | Wang, Y.; Yu, D.-F.; Liu, Y.-Z.; Wei, H.; Luo, Y.-C.; Dixon, D. J.; Xu, P.-F. Chem.–Eur. J. 2010, 16, 3922–3925. doi:10.1002/chem.201000059 |
| 74. | Xu, F.; Corley, E.; Murry, J. A.; Tschaen, D. M. Org. Lett. 2007, 9, 2669–2672. doi:10.1021/ol070909n |
| 55. | Ye, J.; Dixon, D. J.; Hynes, P. S. Chem. Commun. 2005, 4481–4483. doi:10.1039/b508833j |
| 75. | Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2009, 11, 4512–4515. doi:10.1021/ol901640v |
| 76. | Pelletier, S. M.-C.; Ray, P. C.; Dixon, D. J. Org. Lett. 2011, 13, 6406–6409. doi:10.1021/ol202710g |
| 77. | Barber, D. M.; Sanganee, H.; Dixon, D. J. Chem. Commun. 2011, 47, 4379–4381. doi:10.1039/c1cc10751h |
| 78. | Rychnovsky, S. D.; Beauchamp, T.; Vaidyanathan, R.; Kwan, T. J. Org. Chem. 1998, 63, 6363–6374. doi:10.1021/jo9808831 |
| 79. | Westermann, B. Angew. Chem., Int. Ed. 2003, 42, 151–153. doi:10.1002/anie.200390071 |
| 80. | Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626–2704. doi:10.1021/cr100204f |
| 70. | Nara, S.; Tanaka, R.; Eishima, J.; Hara, M.; Takahashi, Y.; Otaki, S.; Foglesong, R. J.; Hughes, P. F.; Turkington, S.; Kanda, Y. J. Med. Chem. 2003, 46, 2467–2473. doi:10.1021/jm020522k |
| 71. | Tanaka, R.; Rubio, A.; Harn, N. K.; Gernert, D.; Grese, T. A.; Eishima, J.; Hara, M.; Yoda, N.; Ohashi, R.; Kuwabara, T.; Soga, S.; Akinaga, S.; Nara, S.; Kanda, Y. Bioorg. Med. Chem. 2007, 15, 1363–1382. doi:10.1016/j.bmc.2006.11.007 |
| 60. | Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389–1391. doi:10.1021/ol800108u |
| 72. | Pei, Z.; Li, X.; von Geldern, T. W.; Longenecker, K.; Pireh, D.; Stewart, K. D.; Backes, B. J.; Lai, C.; Lubben, T. H.; Ballaron, S. J.; Beno, D. W. A.; Kempf-Grote, A. J.; Sham, H. L.; Trevillyan, J. M. J. Med. Chem. 2007, 50, 1983–1987. doi:10.1021/jm061436d |
| 73. | Xu, F.; Corley, E.; Zacuto, M.; Conlon, D. A.; Pipik, B.; Humphrey, G.; Murry, J.; Tschaen, D. J. Org. Chem. 2010, 75, 1343–1353. doi:10.1021/jo902573q |
| 61. | Jakubec, P.; Helliwell, M.; Dixon, D. J. Org. Lett. 2008, 10, 4267–4270. doi:10.1021/ol801666w |
| 62. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. doi:10.1021/ja908399s |
| 63. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037–10039. doi:10.1039/c1cc13665h |
| 64. | Jakubec, P.; Kyle, A. F.; Calleja, J.; Dixon, D. J. Tetrahedron Lett. 2011, 52, 6094–6097. doi:10.1016/j.tetlet.2011.09.016 |
| 65. | Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563 |
© 2012 Jakubec et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)