Abstract
Sensitised photolyses of ethoxycarbonyl oximes of aromatic and heteroaromatic ketones yielded iminyl radicals, which were characterised by EPR spectroscopy. Iminyls with suitably placed arene or heteroarene acceptors underwent cyclisations yielding phenanthridine-type products from ortho-additions. For benzofuran and benzothiophene acceptors, spiro-cyclisation predominated at low temperatures, but thermodynamic control ensured ortho-products, benzofuro- or benzothieno-isoquinolines, formed at higher temperatures. Estimates by steady-state kinetic EPR established that iminyl radical cyclisations onto aromatics took place about an order of magnitude more slowly than prototypical C-centred radicals. The cyclisation energetics were investigated by DFT computations, which gave insights into factors influencing the two cyclisation modes.
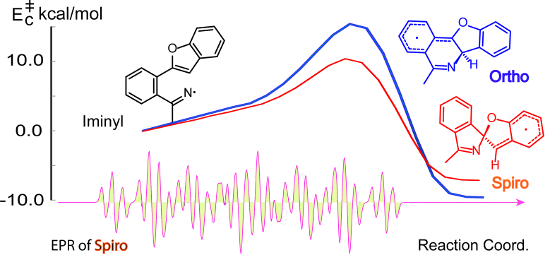
Graphical Abstract
Introduction
Radical cyclisations onto aromatic acceptors take place readily, even though disruption of the 6π-electron system necessarily occurs. The most commonly encountered type is C-centred radical addition (often an aryl radical) to an aromatic or heteroaromatic ring ortho to the point of attachment of the tether. In Pschorr and related processes re-aromatisation follows with production of phenanthrene-type derivatives [1,2]. Spiro-cyclisations in which tethered radicals add to the ipso-C-atoms of the rings are less common, although minor spiro-products not infrequently accompany the main ortho-ones in Pschorr syntheses [3-5]. Cyclisations onto arenes by N-centred radicals are rarer, but iminyl radical ArC(R)=N• closures are well documented. Forrester and co-workers were probably the first to utilise iminyl radicals synthetically. They obtained iminyls by persulfate oxidation of imino-oxyacetic acids in aqueous solvents and prepared azines [6], N-heterocycles [7,8], and other derivatives [9]. This research initiated spiralling interest by synthetic chemists in iminyl radical-mediated preparations. Recently iminyls have been generated from quite a variety of precursors [10-15], and their cyclisations onto arenes [16-21] and heteroarenes [22-24] have attracted attention. Iminyl cyclisations have also been utilised in natural-product syntheses [10,16,25,26]. Iminyl radical spiro-cyclisations onto aromatics have been reported in a few cases, and spiro-intermediates have occasionally been proposed in mechanistic explanations [15,18,27-29].
Although a moderate amount of information about iminyl radical structure and reactivity exists, few conceptual tools to help predict their cyclisation selectivity are available. EPR spectroscopic and other evidence established that iminyl radicals behave as σ-type species with their unpaired electrons in orbitals in the nodal plane of their C=N π-systems [30-32]. This precludes substantial delocalization of the unpaired electron into the ring π-system of aryliminyls. Consequently, strong effects from ring substituents of aryliminyls are not expected to come into play. Small to moderate size iminyl radicals terminate rapidly at diffusion-controlled rates by N–N coupling to give azines [32]. β-Scission reactions yielding nitriles do occur, but are not important at T < ~420 K for aryliminyls or for iminyls with primary alkyl substituents [32]. The rate constants for H-abstraction by iminyls yielding imines are more than an order of magnitude slower than for C-centred analogues [33]. Iminyls undergo 5-exo-ring closures onto alkenes about a factor of 25 more slowly than C-centred analogues [34]. Since ring closure is often in competition with H-abstraction, the comparatively slow H-abstraction by iminyls is important for the success of many heterocycle syntheses.
Spiro-cyclisations of iminyls lead to formation of strained quaternary C-atoms, and the spiro-intermediates have no straightforward reaction channel for return to aromaticity. The process might be reversible, depending on the architecture of the chain and the extent of strain in the spiro-radical. On the other hand ortho-cyclisations can easily be followed by return to aromaticity of the cyclohexadienyl radicals, either by transfer out of the labile H-atom, or by transfer of an electron to a suitable sink with generation of the corresponding carbocation, followed by proton loss. Kinetic or other data to help predict which mode would be favoured for a novel iminyl radical is essentially nonexistent.
We discovered recently that oxime carbonates ArC(R)=N–OC(O)OR’ are clean and convenient precursors for iminyl as well as O-centred radicals [26,35]. Their weak N–O bonds selectively cleave on UV photolysis, particularly when sensitised with 4-methoxyacetophenone (MAP), thus facilitating investigations of the behaviour of both iminyl and alkoxycarbonyloxyl radicals R’OC(O)O•. A distinct advantage of these precursors is that they enable the iminyl radical intermediates to be directly monitored by EPR spectroscopy. We have now prepared a representative set of oxime carbonates with the aim of studying competition between ortho- and spiro-ring closures of the released iminyl radicals. Precursors 1a–f, 2a,b, 3 and 4, consist of O-ethoxycarbonyl derivatives of oximes with various aromatic and heteroaromatic architectures (Figure 1). Compounds 1a–f contain comparatively rigid arms and their aromatic acceptors range from electron-withdrawing to electron-releasing in character. In compounds 2a,b and 3 heteroarenes replace the benzene rings and in 4 the arm is much more flexible.
![[1860-5397-9-120-1]](/bjoc/content/figures/1860-5397-9-120-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: O-Ethoxycarbonyl oximes prepared.
Figure 1: O-Ethoxycarbonyl oximes prepared.
This paper reports our study of the chemistry of these compounds by means of product analyses, solution EPR spectroscopy and DFT computations. We encountered an intriguing interplay between spiro-cyclisation and ortho-cyclisation of the iminyl intermediates and factors affecting this are weighed up.
Results and Discussion
The arene and heteroarene-based oxime carbonates 1 and 2 were prepared as described previously by reaction of the corresponding aromatic ethanone oximes with ethyl chloroformate [26]. Precursors 3 and 4 were made in a similar way from the oximes of 2-furanylphenylethanone and 1,3-diphenylpropan-1-one.
Ring closures of aryl-iminyl radicals
Individual members of the set of substituted biphenyl O-ethoxycarbonyl oximes 1a–f were UV irradiated for 3 h at ambient temperature in deoxygenated benzotrifluoride solutions with 1 equiv wt/wt of MAP as a photosensitizer. As communicated previously, 3-substituted phenanthridines 10a–f were isolated in good to quantitative yields (52–99%, Scheme 1) irrespective of the nature of the 4-substituent [26]. No spiro-products were detected even by GC–MS analyses of reaction mixtures. Byproducts included traces of imines ArC(R2)=NH (ImH) and the ketones ArC(R2)=O from imine hydrolyses. Photolysis of the more flexible precursor 4 gave a complex mixture of products. The MS and NMR data indicated that the main components were probably the corresponding imine ImH and ketone together with the iminyl radical dimer (Im2). Neither spiro- nor ortho-cyclised products had formed. Therefore, for 4 in PhCF3 solvent, iminyl radical ring closures were too slow to compete with H-atom abstractions and terminations.
![[1860-5397-9-120-i1]](/bjoc/content/inline/1860-5397-9-120-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photochemical reactions of biphenyl oxime carbonates.
Scheme 1: Photochemical reactions of biphenyl oxime carbonates.
The photolytic reactions of oxime carbonates 1a–f and 4 were next investigated by 9 GHz EPR spectroscopy. Deaerated samples of each oxime carbonate, plus 1 equiv of MAP, in t-BuPh or cyclopropane solvent, were irradiated with a 500 W unfiltered Hg lamp directly in the spectrometer resonant cavity. The spectrum obtained from precursor 1f (Figure 2) shows a central 1:1:1 triplet from iminyl radical 5f together with a second species. Similar spectra were obtained, in the temperature range 210 to 270 K, from all the other members of the set, including 4, showing the corresponding iminyl radicals plus the same second radical. The EPR parameters of all the iminyls were very similar [g = 2.0030¸ a(N) = 10.0 G] and closely in line with literature data for ArCMe=N• type radicals [36,37].
![[1860-5397-9-120-2]](/bjoc/content/figures/1860-5397-9-120-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: EPR spectrum during photolysis of 1f in t-BuPh at 240 K. Top (black): experimental spectrum. Bottom (red): computer simulation.
Figure 2: EPR spectrum during photolysis of 1f in t-BuPh at 240 K. Top (black): experimental spectrum. Bottom...
Simulation of the second species indicated one large and four smaller doublet hyperfine splittings (hfs) characteristic of a cyclohexadienyl type radical (Table 1). This spectrum was evidently due to the intermediate from addition of some radical meta to the tert-butyl substituent of the solvent. It is known that ArCMe=N• type radicals do not add to t-BuPh under EPR conditions [19,37] neither do EtO• radicals (from dissociation of 6), and hence, we assign this spectrum to the ethoxycarbonyloxyl adduct 9a. This identification was supported by a DFT computation [38] that gave hfs in close agreement with experiment (Table 1). This was a surprising result because previously the only radicals of type 9 that had been spectroscopically detected had resulted from additions of phenyl [34] or bridgehead radicals (bicyclo[2.2.2]oct-1-yl and adamantyl) [39]. These are localized σ-type radicals with bent or pyramidal centres and significant s-character. Ethoxycarbonyloxyl (6) is planar with a SOMO delocalized over the whole OC(O)O unit. The observation of 9a at temperatures below 273 K is dramatic evidence of the exceptionally high reactivity of alkoxycarbonyloxyl radicals. An interesting feature was that addition was selective for meta to the t-Bu group; as was previously observed with the σ-radicals. Product studies with bridgehead radicals at higher temperatures (80 °C) had also revealed this preference for meta-attack [40,41]. The selectivity for meta-addition may result from the electron-releasing character of the t-Bu substituent. The SOMO in 9a has a node at C(3) so electron–electron repulsion is smaller than in the SOMOs for para- or ortho-attack. This will lower the activation energy for meta-addition relative to para- or ortho-addition.
Table 1: EPR parameters of cyclohexadienyl radicals 9 from meta-additions to t-BuPha.
| Radical | T/K or method | g-factor | a(H1) | a(H2) | a(H4) | a(H5) | a(H6) |
|---|---|---|---|---|---|---|---|
| 9a | 240 | 2.0025 | 34.6 | 8.1 | 13.1 | 2.8 | 9.2 |
| 9a | DFTb | – | 35.0 | −8.3 | −12.8 | 3.5 | −9.8 |
| 9bc | 210 | 2.0026 | 33.5 | 8.1 | 13.1 | 2.7 | 9.3 |
| 9(Ph)d | 220 | 2.0030 | 35.5 | 8.1 | 13.3 | 2.7 | 9.1 |
| 9(222)e | 220 | 2.0030 | 42.6 | 8.1 | 13.1 | 2.8 | 9.0 |
aAt 9.4 GHz in t-BuPh solution; hfs in Gauss. Note that the signs of hfs cannot be obtained from isotropic EPR spectra. bUB3LYP/6-311+G(2d,p); hfs computed with the epr-iii basis set [42] designed for EPR hfs, were virtually identical. cR. T. McBurney and J. C. Walton unpublished. dAs 9 but with Ph in place of OC(O)OEt [34]. eAs 9 but with bicyclo[2.2.2]oct-1-yl in place of OC(O)OEt [39].
For all precursors 1a–f the EPR spectra revealed uncyclised iminyls (5a–f) together with 9 up to T ~270 K. Above this temperature the EPR spectra became too weak for radical identification. No cyclohexadienyl type radicals from either spiro or ortho ring closure (7 or 8) were detected. It can be concluded that the iminyl cyclisations are comparatively slow and, based on the previous product analyses, the ortho- (Ar1-6) mode predominates at room temperature and above.
Spiro-cyclisations with benzofuran and benzothiophene acceptors
EPR spectra from oxime carbonate 2a, containing a benzofuran acceptor, gave well resolved spectra only at 230–235 K (Figure 3). The corresponding benzofuranyl-iminyl was not detectable at 230 K or above.
![[1860-5397-9-120-3]](/bjoc/content/figures/1860-5397-9-120-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: EPR spectrum during photolysis of 2a in t-BuPh at 230 K. Top (blue): experiment; bottom (red): simulation.
Figure 3: EPR spectrum during photolysis of 2a in t-BuPh at 230 K. Top (blue): experiment; bottom (red): simu...
The EPR hfs obtained from simulation of the spectrum (Table 2) show this to be a benzyl type radical and we assign it structure 12a (Scheme 2). A DFT computation for 12a at the UB3LYP/6-311+G(2d,p) level of theory gave hfs in close agreement with experiment (Table 2). The EPR hfs are certainly not consistent with structure 13, which was not spectroscopically detected. It is evident therefore that iminyl radical 11a rapidly and selectively undergoes spiro-cyclisation with the benzofuran acceptor.
Table 2: EPR parameters of spiro-benzyl radicals 12 derived from 2a,ba.
| Radical | T/K or DFT method | g-factor | a(Hα) | a(H3) | a(H4) | a(H5) | a(H6) | a(N) |
|---|---|---|---|---|---|---|---|---|
|
12a
(X = O) |
230 | 2.0025 | 14.9 | 1.4 | 6.3 | 1.4 | 5.0 | 5.4 |
|
12a
(X = O) |
UB3LYP/6-311+G(2d,p) | – | −14.2 | 1.6 | −5.9 | 1.6 | −5.0 | 5.2 |
|
12b
(X = S) |
230 | – | 13.0 | 1.5 | 6.0 | 1.5 | 5.3 | 4.1 |
|
12b
(X = S) |
UB3LYP/6-311+G(2d,p) | −13.9 | 2.0 | −5.9 | 2.0 | −5.1 | 5.8 | |
aAt 9.4 GHz in t-BuPh solution; hfs in Gauss.
![[1860-5397-9-120-i2]](/bjoc/content/inline/1860-5397-9-120-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Ring closure of iminyl radicals derived from 2a,b.
Scheme 2: Ring closure of iminyl radicals derived from 2a,b.
Rate constants of ring-closure reactions can be determined for sterically unhindered radicals by measurements of the concentrations of the ring-open and cyclised radicals under EPR conditions [43-45]. Steady-state concentrations of 12a were determined in the usual way from the spectra [46].
We estimated that the concentration ratio [12a]/[11a] > 4 at 230 K, and hence ksc(230 K) > 55 s−1 (see Supporting Information File 1 for details). Most radical cyclisations have Arrhenius log(Ac) ≈ 10.5 s−1 [47,48] and, by assuming that this holds for 12a, an activation energy Esc < 9 kcal mol−1 and ksc(300 K) > 5 × 103 s−1 are obtained (see Supporting Information File 1). The rate constant for 5-exo-cyclisation of the phenylpentenyliminyl radical 15 was reported [34] to be kc(300 K) = 8.8 × 103 s−1 with Ec = 8.3 kcal mol−1 and therefore, particularly in view of the large resonance stabilisation of spiro-cyclised radical 12a, these rate parameters seem to be very reasonable estimates (Table 3).
Table 3: Rate data for spiro- and other cyclisations of C- and N-centred radicals.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-120-i3.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-120-i4.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-120-i5.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-120-i6.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-120-i7.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-120-i8.svg?max-width=637&scale=1.0)
aValues in parenthesis assumed.
Curiously, analysis of the products from a photolysis of 2a carried out at higher temperature (rt) in benzotrifluoride solvent, showed benzofuro[3,2-c]isoquinoline derivative 14a to be the main product (65%) [26]. This implied ortho-radicals 13 as intermediates and appeared to conflict with the EPR result. The most likely explanation is that, at the temperature of the preparative experiments (~100 K higher than the EPR study) the spiro-cyclisation is reversible whereas the 6-ortho-process is not. The ortho-product then accumulates because of thermodynamic control. Alternatively, spiro-radical 12a might rearrange via a tetracyclic aziridinyl intermediate (or transition state) at higher temperatures. DFT computations (see below) undermined this possibility however.
EPR experiments with the benzothiophene-containing precursor 2b showed a complex spectrum from at least two radicals. A reasonable simulation (see Supporting Information File 1) was obtained as a combination of the spiro-radical 12b (hfs in Table 2) and the solvent-derived adduct 9a. Most likely therefore spiro-cyclisation predominates at low temperature but again thermodynamic control takes over at higher temperatures because 14b was isolated as the main product.
EPR spectra from the furan-containing precursor 3, and from the more flexible aromatic precursor 4, revealed only the corresponding ring-open iminyl radicals and neither spiro- nor ortho-radicals in the temperature range 230–260 K. It can be concluded that the rates of their iminyl ring closures are significantly slower. This accords with expectation, because the cyclised radical from 3, without the benzo-ring of 12, would be less thermodynamically stabilised. The iminyl radical from 4 has a more flexible chain and this probably accounts for its slower ring closure.
Kinetic data for iminyl radical ring closures is compared with analogous data for model C-centred radicals in Table 3. As mentioned above, biphenyl-iminyl radicals 5 were spectroscopically detectable at 270 K; well above the temperature at which 12a underwent spiro-cyclisation. It follows that kc(300 K) for 5b must be < kc for 12a and this information is included in Table 3.
The rate constant for 5-exo-cyclisation the archetype iminyl 15 (Table 3, entry 2) is more than an order of magnitude smaller than the rate constant for hex-5-enyl (16, Table 3, entry 1) [49]. Our rates for ortho- (Table 3, entry 5) and spiro- (Table 3, entry 6) cyclisations of iminyls onto aromatics were also slower than for C-centred radicals (Table 3, entries 3 and 4) [50,51]. The pattern of slower cyclisations for iminyls compared with alkyls seems established for both alkene- and arene-type acceptors.
QM Computations
To shed further light on the spiro versus ortho alternatives we computed the activation parameters [ΔE‡298] and reaction enthalpies [ΔH298], corrected to 298 K for thermal effects, for the spiro- and ortho-cyclisation modes of a representative set of aryliminyl radicals (Table 4). It was known that the DFT UB3LYP/6-31+G(d) and UB3LYP/6-311+G(2d,p) methods gave results in reasonable harmony with experiment for related iminyl radicals [34] and therefore these methods were adopted. For each process the two basis sets gave results in reasonable agreement (Table 4). The cyclisations of the flexible iminyl radicals 4Im (Tabel 4, entries 1 and 2) were predicted to have the highest activation energies in either mode and to be strongly endothermic. This accords well with the absence of cyclised species in the EPR spectra and with the lack of cyclised products.
Table 4: DFT computed activation energies (ΔE‡298) and reaction enthalpies (ΔH298) in kcal mol−1 for aromatic iminyl radicals.
| Entry | Iminyl | Product | Methoda | ΔE‡298 | ΔH298 |
|---|---|---|---|---|---|
| 1 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-120-i9.svg?max-width=637&scale=1.0)
4Im |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-120-i10.svg?max-width=637&scale=1.0)
4sp |
A
B |
16.6
16.9 |
6.6
7.3 |
| 2 | 4Im |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-120-i11.svg?max-width=637&scale=1.0)
4or |
B | 19.9 | 8.0 |
| 3 | 4Im |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-120-i12.svg?max-width=637&scale=1.0)
4az |
B | 32.9 | |
| 4 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-120-i13.svg?max-width=637&scale=1.0)
5a |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-120-i14.svg?max-width=637&scale=1.0)
7a |
A
B |
14.7
15.0 |
7.0
7.6 |
| 5 | 5a |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-120-i15.svg?max-width=637&scale=1.0)
8a |
A
B |
11.3
11.6 |
−2.8
−2.1 |
| 6 | 5a |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-120-i16.svg?max-width=637&scale=1.0)
5az |
B | 29.5 | |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-120-i17.svg?max-width=637&scale=1.0)
3Im |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-120-i18.svg?max-width=637&scale=1.0)
3sp |
A
B |
11.6
12.1 |
−1.1
−0.5 |
| 8 | 3Im |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-120-i19.svg?max-width=637&scale=1.0)
3en |
A
B |
14.2
14.7 |
−2.3
−1.8 |
| 9 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-120-i20.svg?max-width=637&scale=1.0)
11a |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-120-i21.svg?max-width=637&scale=1.0)
12a |
A
B |
8.9
9.3 |
−7.8
−6.9 |
| 10 | 11a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-120-i22.svg?max-width=637&scale=1.0)
13a |
A
B |
12.6
13.1 |
−8.6
−8.1 |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-120-i23.svg?max-width=637&scale=1.0)
11b |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-120-i24.svg?max-width=637&scale=1.0)
12b |
B | 9.5 | −6.2 |
| 12 | 11b |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-120-i25.svg?max-width=637&scale=1.0)
13b |
B | 10.5 | −11.3 |
aA = UB3LYP/6-31+G(d), B = UB3LYP/6-311+G(2d,p).
For the parent biphenyliminyl radical 5a the ortho-ring closure (Table 4, entry 5) was computed to have a lower activation energy than spiro-cyclisation (Table 4, entry 4) and to be exothermic in comparison with the endothermic spiro-mode. This was in good accordance with the observed exclusive formation of phenanthridines derived from radicals 5a–f for these compounds. The possibility of the rearrangement of spiro- to ortho-species via aziridinyl intermediates (or transition states) (az) which contain benzyl stabilization was examined. However, the computations for 4az (Table 4, entry 3) and 5az (Table 4, entry 6) found these processes to be strongly endothermic, so they can probably be ruled out of consideration. Predicted activation energies and reaction enthalpies for spiro- (Table 4, entry 7) and ortho-ring closure (Table 4, entry 8) of the furanyl-iminyl 3Im, suggested spiro-closure would be favoured. However, the magnitude of ΔE‡298 is close to that for ortho-closure of the biphenyl analogue 5a. Thus cyclised products might be obtainable from photolyses of 3 at higher temperatures.
For the benzofuranyl- and benzothiophenyl-iminyl radicals 11a and 11b (Table 4, entries 9–12) the computations predicted a reversal in the preferred mode of ring closure: ΔE‡298 values for spiro-cyclisation were significantly lower than for ortho-cyclisation and lower than for 5a. This agreed well with the EPR detection of the spiro-intermediates 12a and 12b. From the EPR experiments the Esc of < 9 kcal mol−1 for spiro-cyclisation of 11a (see above Table 3) was close to the computed ΔE‡298 values of 8.9 and 9.3 kcal mol−1 (Table 4, entry 9). For both 11a and 11b, although the computed ΔE‡298 values for ortho-ring closure are significantly higher than for spiro-ring closure, the ortho-processes are computed to be more exothermic. Thus, theory supports the idea, derived from the isolation of products from ortho-closure of 14a,b, that thermodynamic control supervenes at higher temperatures.
Some clues as to why the biphenyl-iminyls 5 prefer the ortho mode whereas the benzofuranyl- and benzothiophenyl-iminyls 11 prefer the spiro mode can be obtained by consideration of the structures of the transition states (TS, Figure 4). The top line shows the computed spin density (ρ) maps for the reactant iminyls. In agreement with the EPR spectra, the spin density is largely concentrated in orbitals in the plane of the iminyl units. The middle line of Figure 4 shows the TS structures and SOMOs for spiro-cyclisations of 5a and 11a. For both radicals the iminyl units approach the acceptor rings from vertically above; the angle between the ArC(R)=N unit and the plane of the acceptor ring is ~90° for both TSs. This ensures that overlap between the incoming iminyl orbital and the acceptor ring orbitals is optimum. The bottom line shows the TS structures and SOMOs for the ortho-cyclisations of 5a and 11a. For both radicals the architectures oblige the iminyl units to approach the acceptor rings at much more oblique angles. Thus, overlap of the in-plane iminyl orbital with the acceptor ring orbitals is poorer than for spiro-cyclisation. However, this is offset by the greater resonance delocalisation in the TS frontier orbitals for ortho-cyclisation (see Figure 4). There is a trade-off between these two factors and the default is that the resonance delocalisation factor in ortho-cyclisation prevails. Evidently 11a,b, are exceptional in that the iminyl/acceptor overlap factor outweighs the resonance delocalisation factor.
![[1860-5397-9-120-4]](/bjoc/content/figures/1860-5397-9-120-4.png?scale=1.4&max-width=1024&background=FFFFFF)
Figure 4: DFT computed structures for 5a, 11a and their cyclisation transition states (TS). Top line: spin density distribution maps for iminyls 5a and 11a. Middle line: spiro TSs, r(N···C) for 5a = 1.928 and for 11a = 2.064 Å. Bottom line: ortho TSs, r(N···C) for 5a = 1.954 and for 11a = 2.027 Å.
Figure 4: DFT computed structures for 5a, 11a and their cyclisation transition states (TS). Top line: spin de...
Conclusion
Ethoxycarbonyl oximes of aromatic and heteroaromatic ketones are easily made and handled and have long shelf lives. We have found them to be outstanding precursors for the photochemical generation of aromatic iminyl radicals. Product analyses, EPR spectroscopic observations and DFT computations all converged in indicating that the “default” ring closure mode for iminyl radicals is ortho-cyclisation onto suitably situated aromatic rings. The exception to this rule was spiro-cyclisation onto benzofuran and benzothiophene rings at low temperatures. Even for these acceptors, however, thermodynamic control ensured that the only isolable products were from ortho-closures. Rate parameters for the spiro-cyclisation of 11a, estimated by the steady-state kinetic EPR method, showed the process to be about an order of magnitude slower than for archetype C-centred radicals. This was in harmony with the generally slower reaction rates of iminyl radicals compared with alkyl radicals. DFT-computed energetics were in good agreement with experiment and supported the idea of thermodynamic control for the cyclisation of 11a,b.
Experimental
EPR spectra were obtained at X-Band on Bruker EMX 10/12 spectrometers at St. Andrews and Manchester. Oxime carbonates (2 to 15 mg) and MAP (1 equiv wt/wt) in t-BuPh or benzene (0.5 mL) were prepared in quartz tubes and deaerated by bubbling with nitrogen for 15 min. Photolysis in the resonant cavity was by unfiltered light from a 500 W super-pressure mercury arc lamp or, in the Manchester experiments, from a Luxtel CL300BUV lamp. EPR signals were digitally filtered and double integrated by using the Bruker WinEPR software, and radical concentrations were calculated by reference to the double integral of the signal from a known concentration of the stable radical DPPH run under identical conditions. The majority of EPR spectra were recorded with 2.0 mW power, 0.8 Gpp modulation intensity and gain of 106.
QM calculations were carried out by using the Gaussian 09 program package. Geometries were fully optimised for all model compounds. Optimised structures were characterised as minima or saddle points by frequency calculations. The experimental kinetic and spectroscopic data was all obtained in the nonpolar hydrocarbon solvents t-BuPh or cyclopropane. Solvent effects, particularly differences in solvation between the neutral reactants and neutral transition states, were therefore expected to be minimal. In view of this, no attempt was made to model the effect of the solvent computationally.
Supporting Information
| Supporting Information File 1: General procedures. Preparation and characterization data of oxime carbonates. Sample EPR spectra and kinetic data. 1H and 13C NMR spectra for novel compounds. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Pratsch, G.; Heinrich, M. R. Modern Development in Aryl Radical Chemistry. Radicals in Synthesis III; Topics in Currrent Chemistry, Vol. 320; Springer Verlag: Berlin, Germany, 2012; pp 33–59. doi:10.1007/128_2011_127
Return to citation in text: [1] -
Laali, K. K.; Shokouhimehr, M. Curr. Org. Synth. 2009, 6, 193–202. doi:10.2174/157017909788167275
Return to citation in text: [1] -
Crich, D.; Hwang, J.-T. J. Org. Chem. 1998, 63, 2765–2770. doi:10.1021/jo972197s
Return to citation in text: [1] -
Maggio, B.; Daidone, G.; Raffa, D.; Plescia, S.; Bombieri, G.; Meneghetti, F. Helv. Chim. Acta 2005, 88, 2272–2281. doi:10.1002/hlca.200590161
Return to citation in text: [1] -
Moorthy, J. N.; Samanta, S. J. Org. Chem. 2007, 72, 9786–9789. doi:10.1021/jo7017872
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Meyer, C. J.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 606–611. doi:10.1039/P19790000606
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 612–615. doi:10.1039/P19790000612
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 616–620. doi:10.1039/P19790000616
Return to citation in text: [1] -
Forrester, A. R.; Napier, R. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1981, 984–987. doi:10.1039/P19810000984
Return to citation in text: [1] -
Zard, S. Z. Synlett 1996, 1148–1154. doi:10.1055/s-1996-5698
Return to citation in text: [1] [2] -
Gagosz, F.; Zard, S. Z. Synlett 1999, 1978–1980. doi:10.1055/s-1999-2971
Return to citation in text: [1] -
Bowman, W. R.; Bridge, C. F.; Brookes, P. Tetrahedron Lett. 2000, 41, 8989–8994. doi:10.1016/S0040-4039(00)01596-3
Return to citation in text: [1] -
McCarroll, A. J.; Walton, J. C. Chem. Commun. 2000, 351–352. doi:10.1039/a910346p
Return to citation in text: [1] -
Bencivenni, G.; Lanza, T.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Zanardi, G. J. Org. Chem. 2008, 73, 4721–4724. doi:10.1021/jo800453z
Return to citation in text: [1] -
Lanza, T.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Zanardi, G. Angew. Chem., Int. Ed. 2008, 47, 9439–9442. doi:10.1002/anie.200804333
Return to citation in text: [1] [2] -
Bowman, W. R.; Bridge, C. F.; Cloonan, M. O.; Leach, D. C. Synlett 2001, 765–768. doi:10.1055/s-2001-14592
Return to citation in text: [1] [2] -
Blake, J. A.; Pratt, D. A.; Lin, S.; Walton, J. C.; Mulder, P.; Ingold, K. U. J. Org. Chem. 2004, 69, 3112–3120. doi:10.1021/jo049927y
Return to citation in text: [1] -
Bowman, W. R.; Cloonan, M. O.; Fletcher, A. J.; Stein, T. Org. Biomol. Chem. 2005, 3, 1460–1467. doi:10.1039/b501509j
Return to citation in text: [1] [2] -
Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. Chem. Commun. 2008, 2935–2937. doi:10.1039/b803630f
Return to citation in text: [1] [2] -
Alonso, R.; Campos, P. J.; García, B.; Rodríguez, M. A. Org. Lett. 2006, 8, 3521–3523. doi:10.1021/ol061258i
Return to citation in text: [1] -
Portela-Cubillo, F.; Lymer, J.; Scanlan, E. M.; Scott, J. S.; Walton, J. C. Tetrahedron 2008, 64, 11908–11916. doi:10.1016/j.tet.2008.08.112
Return to citation in text: [1] -
Beaume, A.; Courillon, C.; Derat, E.; Malacria, M. Chem.–Eur. J. 2008, 14, 1238–1252. doi:10.1002/chem.200700884
Return to citation in text: [1] -
Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. J. Org. Chem. 2008, 73, 5558–5565. doi:10.1021/jo800847h
Return to citation in text: [1] -
Alonso, R.; Caballero, A.; Campos, P. J.; Rodríguez, M. A. Tetrahedron 2010, 66, 8828–8831. doi:10.1016/j.tet.2010.09.078
Return to citation in text: [1] -
Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. J. Org. Chem. 2009, 74, 4934–4942. doi:10.1021/jo900629g
Return to citation in text: [1] -
McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a
Return to citation in text: [1] [2] [3] [4] [5] -
Sridar, V.; Babu, G. Synth. Commun. 1997, 27, 323–330. doi:10.1080/00397919708005035
Return to citation in text: [1] -
Calestani, G.; Leardini, R.; McNab, H.; Nanni, D.; Zanardi, G. J. Chem. Soc., Perkin Trans. 1 1998, 1813–1824. doi:10.1039/A800868J
Return to citation in text: [1] -
Bowman, W. R.; Bridge, C. F.; Brookes, P.; Cloonan, M. O.; Leach, D. C. J. Chem. Soc., Perkin Trans. 1 2002, 58–68. doi:10.1039/B108323F
Return to citation in text: [1] -
Neta, P.; Fessenden, R. W. J. Phys. Chem. 1970, 74, 3362–3365. doi:10.1021/j100712a009
Return to citation in text: [1] -
Hudson, R. F.; Lawson, A. J.; Lucken, E. A. C. J. Chem. Soc. D 1971, 807–808. doi:10.1039/C29710000807
Return to citation in text: [1] -
Griller, D.; Mendenhall, G. D.; Van Hoof, W.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 6068–6070. doi:10.1021/ja00826a018
Return to citation in text: [1] [2] [3] -
Le Tadic-Biadatti, M.-H.; Callier-Dublanchet, A.-C.; Horner, J. H.; Quiclet-Sire, B.; Zard, S. Z.; Newcomb, M. J. Org. Chem. 1997, 62, 559–563. doi:10.1021/jo961530+
Return to citation in text: [1] -
Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902
Return to citation in text: [1] [2] [3] [4] [5] [6] -
McBurney, R. T.; Harper, A. D.; Slawin, A. M. Z.; Walton, J. C. Chem. Sci. 2012, 3, 3436–3444. doi:10.1039/c2sc21298f
Return to citation in text: [1] -
Hudson, R. F.; Record, K. A. F. J. Chem. Soc., Chem. Commun. 1976, 831–832. doi:10.1039/C39760000831
Return to citation in text: [1] -
McCarroll, A. J.; Walton, J. C. J. Chem. Soc., Perkin Trans. 2 2000, 2399–2409. doi:10.1039/b007212p
Return to citation in text: [1] [2] -
Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Binmore, G. T.; Walton, J. C.; Adcock, W.; Clark, C. I.; Kristic, A. R. Magn. Reson. Chem. 1995, 33, S53–S59. doi:10.1002/mrc.1260331310
Return to citation in text: [1] [2] -
Mangini, A.; Spagnolo, P.; Tassi, D.; Tiecco, M.; Zanirato, P. Tetrahedron 1972, 28, 3485–3488. doi:10.1016/0040-4020(72)88109-2
Return to citation in text: [1] -
Tiecco, M. Pure Appl. Chem. 1981, 53, 239–258. doi:10.1351/pac198153010239
Return to citation in text: [1] -
Barone, V. Structure, Magnetic Properties and Reactivities of Open-Shell Species From Density Functional and Self-Consistent Hybrid Methods. In Recent Advances in Density Functional Methods; Chong, D. P., Ed.; Recent Advances in Computational Chemistry, Vol. 1; World Scientific: Singapore, 1995; pp 287–334. doi:10.1142/9789812830586_0008
Return to citation in text: [1] -
Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 193–200. doi:10.1021/ar50151a001
Return to citation in text: [1] -
Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317–323. doi:10.1021/ar50153a004
Return to citation in text: [1] -
Walton, J. C. J. Chem. Soc., Perkin Trans. 2 1987, 231–235. doi:10.1039/p29870000231
Return to citation in text: [1] -
Walton, J. C. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; John Wiley & Sons: Chichester, UK, 2012; pp 147–174.
Return to citation in text: [1] -
Beckwith, A. L. J.; Brumby, S. In Landolt-Börnstein, Radical Reaction Rates in Liquids; Fischer, H., Ed.; Springer Verlag: Berlin, 1994; Vol. II18a, pp 171–254.
Return to citation in text: [1] -
Beckwith, A. L. J. In Landolt-Börnstein, Radical Reaction Rates in Liquids; Fischer, H., Ed.; Springer Verlag: Berlin, 1984; Vol. II13a, pp 252–315.
Return to citation in text: [1] -
Chatgilialoglu, C.; Ingold, K. U.; Scaiano, J. C. J. Am. Chem. Soc. 1981, 103, 7739–7742. doi:10.1021/ja00416a008
Return to citation in text: [1] [2] -
Julia, M. Pure Appl. Chem. 1974, 40, 553–567. doi:10.1351/pac197440040553
Return to citation in text: [1] [2] -
Kochi, J. K.; Gilliom, R. D. J. Am. Chem. Soc. 1964, 86, 5251–5256. doi:10.1021/ja01077a042
Return to citation in text: [1] [2]
| 43. | Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 193–200. doi:10.1021/ar50151a001 |
| 44. | Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317–323. doi:10.1021/ar50153a004 |
| 45. | Walton, J. C. J. Chem. Soc., Perkin Trans. 2 1987, 231–235. doi:10.1039/p29870000231 |
| 46. | Walton, J. C. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; John Wiley & Sons: Chichester, UK, 2012; pp 147–174. |
| 47. | Beckwith, A. L. J.; Brumby, S. In Landolt-Börnstein, Radical Reaction Rates in Liquids; Fischer, H., Ed.; Springer Verlag: Berlin, 1994; Vol. II18a, pp 171–254. |
| 48. | Beckwith, A. L. J. In Landolt-Börnstein, Radical Reaction Rates in Liquids; Fischer, H., Ed.; Springer Verlag: Berlin, 1984; Vol. II13a, pp 252–315. |
| 1. | Pratsch, G.; Heinrich, M. R. Modern Development in Aryl Radical Chemistry. Radicals in Synthesis III; Topics in Currrent Chemistry, Vol. 320; Springer Verlag: Berlin, Germany, 2012; pp 33–59. doi:10.1007/128_2011_127 |
| 2. | Laali, K. K.; Shokouhimehr, M. Curr. Org. Synth. 2009, 6, 193–202. doi:10.2174/157017909788167275 |
| 9. | Forrester, A. R.; Napier, R. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1981, 984–987. doi:10.1039/P19810000984 |
| 34. | Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902 |
| 49. | Chatgilialoglu, C.; Ingold, K. U.; Scaiano, J. C. J. Am. Chem. Soc. 1981, 103, 7739–7742. doi:10.1021/ja00416a008 |
| 7. | Forrester, A. R.; Gill, M.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 612–615. doi:10.1039/P19790000612 |
| 8. | Forrester, A. R.; Gill, M.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 616–620. doi:10.1039/P19790000616 |
| 26. | McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a |
| 35. | McBurney, R. T.; Harper, A. D.; Slawin, A. M. Z.; Walton, J. C. Chem. Sci. 2012, 3, 3436–3444. doi:10.1039/c2sc21298f |
| 50. | Julia, M. Pure Appl. Chem. 1974, 40, 553–567. doi:10.1351/pac197440040553 |
| 51. | Kochi, J. K.; Gilliom, R. D. J. Am. Chem. Soc. 1964, 86, 5251–5256. doi:10.1021/ja01077a042 |
| 6. | Forrester, A. R.; Gill, M.; Meyer, C. J.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 606–611. doi:10.1039/P19790000606 |
| 32. | Griller, D.; Mendenhall, G. D.; Van Hoof, W.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 6068–6070. doi:10.1021/ja00826a018 |
| 51. | Kochi, J. K.; Gilliom, R. D. J. Am. Chem. Soc. 1964, 86, 5251–5256. doi:10.1021/ja01077a042 |
| 3. | Crich, D.; Hwang, J.-T. J. Org. Chem. 1998, 63, 2765–2770. doi:10.1021/jo972197s |
| 4. | Maggio, B.; Daidone, G.; Raffa, D.; Plescia, S.; Bombieri, G.; Meneghetti, F. Helv. Chim. Acta 2005, 88, 2272–2281. doi:10.1002/hlca.200590161 |
| 5. | Moorthy, J. N.; Samanta, S. J. Org. Chem. 2007, 72, 9786–9789. doi:10.1021/jo7017872 |
| 33. | Le Tadic-Biadatti, M.-H.; Callier-Dublanchet, A.-C.; Horner, J. H.; Quiclet-Sire, B.; Zard, S. Z.; Newcomb, M. J. Org. Chem. 1997, 62, 559–563. doi:10.1021/jo961530+ |
| 26. | McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a |
| 10. | Zard, S. Z. Synlett 1996, 1148–1154. doi:10.1055/s-1996-5698 |
| 16. | Bowman, W. R.; Bridge, C. F.; Cloonan, M. O.; Leach, D. C. Synlett 2001, 765–768. doi:10.1055/s-2001-14592 |
| 25. | Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. J. Org. Chem. 2009, 74, 4934–4942. doi:10.1021/jo900629g |
| 26. | McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a |
| 30. | Neta, P.; Fessenden, R. W. J. Phys. Chem. 1970, 74, 3362–3365. doi:10.1021/j100712a009 |
| 31. | Hudson, R. F.; Lawson, A. J.; Lucken, E. A. C. J. Chem. Soc. D 1971, 807–808. doi:10.1039/C29710000807 |
| 32. | Griller, D.; Mendenhall, G. D.; Van Hoof, W.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 6068–6070. doi:10.1021/ja00826a018 |
| 34. | Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902 |
| 22. | Beaume, A.; Courillon, C.; Derat, E.; Malacria, M. Chem.–Eur. J. 2008, 14, 1238–1252. doi:10.1002/chem.200700884 |
| 23. | Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. J. Org. Chem. 2008, 73, 5558–5565. doi:10.1021/jo800847h |
| 24. | Alonso, R.; Caballero, A.; Campos, P. J.; Rodríguez, M. A. Tetrahedron 2010, 66, 8828–8831. doi:10.1016/j.tet.2010.09.078 |
| 32. | Griller, D.; Mendenhall, G. D.; Van Hoof, W.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 6068–6070. doi:10.1021/ja00826a018 |
| 16. | Bowman, W. R.; Bridge, C. F.; Cloonan, M. O.; Leach, D. C. Synlett 2001, 765–768. doi:10.1055/s-2001-14592 |
| 17. | Blake, J. A.; Pratt, D. A.; Lin, S.; Walton, J. C.; Mulder, P.; Ingold, K. U. J. Org. Chem. 2004, 69, 3112–3120. doi:10.1021/jo049927y |
| 18. | Bowman, W. R.; Cloonan, M. O.; Fletcher, A. J.; Stein, T. Org. Biomol. Chem. 2005, 3, 1460–1467. doi:10.1039/b501509j |
| 19. | Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. Chem. Commun. 2008, 2935–2937. doi:10.1039/b803630f |
| 20. | Alonso, R.; Campos, P. J.; García, B.; Rodríguez, M. A. Org. Lett. 2006, 8, 3521–3523. doi:10.1021/ol061258i |
| 21. | Portela-Cubillo, F.; Lymer, J.; Scanlan, E. M.; Scott, J. S.; Walton, J. C. Tetrahedron 2008, 64, 11908–11916. doi:10.1016/j.tet.2008.08.112 |
| 34. | Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902 |
| 10. | Zard, S. Z. Synlett 1996, 1148–1154. doi:10.1055/s-1996-5698 |
| 11. | Gagosz, F.; Zard, S. Z. Synlett 1999, 1978–1980. doi:10.1055/s-1999-2971 |
| 12. | Bowman, W. R.; Bridge, C. F.; Brookes, P. Tetrahedron Lett. 2000, 41, 8989–8994. doi:10.1016/S0040-4039(00)01596-3 |
| 13. | McCarroll, A. J.; Walton, J. C. Chem. Commun. 2000, 351–352. doi:10.1039/a910346p |
| 14. | Bencivenni, G.; Lanza, T.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Zanardi, G. J. Org. Chem. 2008, 73, 4721–4724. doi:10.1021/jo800453z |
| 15. | Lanza, T.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Zanardi, G. Angew. Chem., Int. Ed. 2008, 47, 9439–9442. doi:10.1002/anie.200804333 |
| 15. | Lanza, T.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Zanardi, G. Angew. Chem., Int. Ed. 2008, 47, 9439–9442. doi:10.1002/anie.200804333 |
| 18. | Bowman, W. R.; Cloonan, M. O.; Fletcher, A. J.; Stein, T. Org. Biomol. Chem. 2005, 3, 1460–1467. doi:10.1039/b501509j |
| 27. | Sridar, V.; Babu, G. Synth. Commun. 1997, 27, 323–330. doi:10.1080/00397919708005035 |
| 28. | Calestani, G.; Leardini, R.; McNab, H.; Nanni, D.; Zanardi, G. J. Chem. Soc., Perkin Trans. 1 1998, 1813–1824. doi:10.1039/A800868J |
| 29. | Bowman, W. R.; Bridge, C. F.; Brookes, P.; Cloonan, M. O.; Leach, D. C. J. Chem. Soc., Perkin Trans. 1 2002, 58–68. doi:10.1039/B108323F |
| 49. | Chatgilialoglu, C.; Ingold, K. U.; Scaiano, J. C. J. Am. Chem. Soc. 1981, 103, 7739–7742. doi:10.1021/ja00416a008 |
| 36. | Hudson, R. F.; Record, K. A. F. J. Chem. Soc., Chem. Commun. 1976, 831–832. doi:10.1039/C39760000831 |
| 37. | McCarroll, A. J.; Walton, J. C. J. Chem. Soc., Perkin Trans. 2 2000, 2399–2409. doi:10.1039/b007212p |
| 26. | McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a |
| 34. | Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902 |
| 26. | McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a |
| 34. | Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902 |
| 39. | Binmore, G. T.; Walton, J. C.; Adcock, W.; Clark, C. I.; Kristic, A. R. Magn. Reson. Chem. 1995, 33, S53–S59. doi:10.1002/mrc.1260331310 |
| 40. | Mangini, A.; Spagnolo, P.; Tassi, D.; Tiecco, M.; Zanirato, P. Tetrahedron 1972, 28, 3485–3488. doi:10.1016/0040-4020(72)88109-2 |
| 41. | Tiecco, M. Pure Appl. Chem. 1981, 53, 239–258. doi:10.1351/pac198153010239 |
| 42. | Barone, V. Structure, Magnetic Properties and Reactivities of Open-Shell Species From Density Functional and Self-Consistent Hybrid Methods. In Recent Advances in Density Functional Methods; Chong, D. P., Ed.; Recent Advances in Computational Chemistry, Vol. 1; World Scientific: Singapore, 1995; pp 287–334. doi:10.1142/9789812830586_0008 |
| 34. | Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J. C. J. Phys. Chem. A 2009, 113, 10005–10012. doi:10.1021/jp9047902 |
| 39. | Binmore, G. T.; Walton, J. C.; Adcock, W.; Clark, C. I.; Kristic, A. R. Magn. Reson. Chem. 1995, 33, S53–S59. doi:10.1002/mrc.1260331310 |
| 19. | Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. Chem. Commun. 2008, 2935–2937. doi:10.1039/b803630f |
| 37. | McCarroll, A. J.; Walton, J. C. J. Chem. Soc., Perkin Trans. 2 2000, 2399–2409. doi:10.1039/b007212p |
© 2013 McBurney and Walton; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)