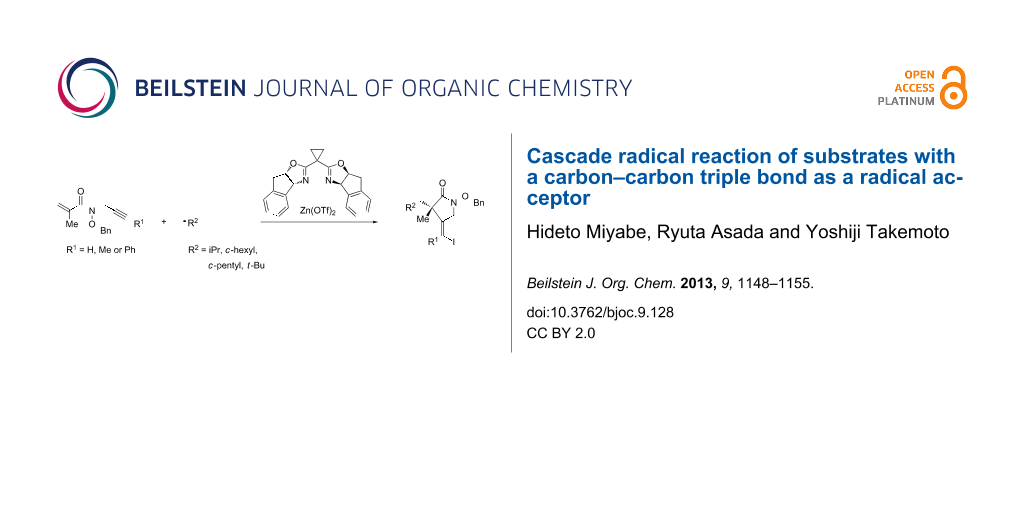
Abstract
The limitation of hydroxamate ester as a chiral Lewis acid coordination moiety was first shown in an intermolecular reaction involving a radical addition and sequential allylation processes. Next, the effect of hydroxamate ester was studied in the cascade addition–cyclization–trapping reaction of substrates with a carbon–carbon triple bond as a radical acceptor. When substrates with a methacryloyl moiety and a carbon–carbon triple bond as two polarity-different radical acceptors were employed, the cascade reaction proceeded effectively. A high level of enantioselectivity was also obtained by a proper combination of chiral Lewis acid and these substrates.

Graphical Abstract
Introduction
Strategies involving a cascade process offer the advantage of multiple carbon–carbon and/or carbon–heteroatom bond formations in a single operation. Radical chemistry has been developed as one of the most powerful tools for carbon–carbon bond formation in organic synthesis [1-20]. Particularly, the advantages for utilizing the radical methodologies are the high functional group tolerance and the mild reaction conditions, because radical intermediates are not charged species. Therefore, a number of extensive investigations into sequential radical reactions have been reported over the past fifteen years and significant progress has been made in recent years [21-36]. We have also directed our efforts toward the development of new and efficient cascade approaches for the construction of carbon–carbon/heteroatom bonds based on radical chemistry. These approaches can be classified into two categories according to their reaction mechanism (Figure 1) [37-43].
![[1860-5397-9-128-1]](/bjoc/content/figures/1860-5397-9-128-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cascade bond formation based on radical reactions.
Figure 1: Cascade bond formation based on radical reactions.
Enantioselective radical reactions have been intensively studied over the past fifteen years. Compared with stereocontrol studies on intermolecular radical reactions, the enantioselective stereocontrol in radical cyclizations still remains a major challenge [44-68]. We have also investigated a new type of chiral Lewis acid mediated cyclization approach for cascade bond-forming reactions via sequential radical–radical processes (Figure 2) [39-43]. In these studies, the control of the enantioselectivities was achieved by the introduction of a hydroxamate ester as a two-point-binding coordination tether into the middle of substrates A, together with the control of the rotamer population of substrates [39,42]. In this paper, we describe in detail the cascade addition–cyclization–trapping reaction of substrates with a carbon–carbon triple bond as a radical acceptor as well as the effect of hydroxamate ester as a Lewis acid coordination moiety. Some results have been reported in our preliminary communication [39].
![[1860-5397-9-128-2]](/bjoc/content/figures/1860-5397-9-128-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Our method for controlling the geometry of substrates and the stereochemistry of cyclization.
Figure 2: Our method for controlling the geometry of substrates and the stereochemistry of cyclization.
Results and Discussion
Renaud’s group showed in 2002 that hydroxamic acid derivatives are useful achiral templates in enantioselective Diels–Alder reactions [69,70]. To study the effect of hydroxamate ester as an achiral template in the intermolecular radical reaction, our experiments began with the investigation of cascade radical addition–allylation of hydroxamate esters 3A–C having an acryloyl moiety (Scheme 1). The reactions were evaluated in CH2Cl2 at −78 °C by employing isopropyl iodide, allyltin reagent, and Et3B as a radical initiator. The enantiomeric purities of products were checked by chiral HPLC analysis. The effect of the substituents R1 and R2 of hydroxamate esters 3A–C on yield and selectivity was evaluated in the presence of a chiral Lewis acid prepared from box ligand L1 and Zn(OTf)2. The results are shown in Scheme 1. Although good enantioselectivities were not observed, the size of the substituents had an impact on enantioselectivity with the larger group leading to lower ee. These observations indicate that the formation of the rigid ternary complex of hydroxamate ester, Zn(OTf)2 and the ligand L1 is required for enantioselective transformation. A similar trend was observed in our studies on the addition–cyclization–trapping reaction of hydroxamate esters [39,42]. The chiral Lewis acid promoted the reaction of substrate 3A having a bulky 2-naphthylmethyl group as substituent R2 to form the product 4A in 40% yield with 7% ee. Moderate enantioselectivity was observed by employing the substrate 3B having a benzyl group as R1 and a methyl group as R2. Particularly, the steric factor of the fluxional substituent R1 affected not only enantioselectivity but also the chemical efficiency. The use of 3C having a 2-naphthylmethyl group as R1 led to a decrease in the chemical yield, probably because of the steric repulsion by a bulky substituent R1 leading to the dissociation of the chiral Lewis acid. In these studies, the absolute configuration at newly generated stereocenters has been not determined.
![[1860-5397-9-128-i1]](/bjoc/content/inline/1860-5397-9-128-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Effect of hydroxamate ester on intermolecular C–C bond-forming reactions.
Scheme 1: Effect of hydroxamate ester on intermolecular C–C bond-forming reactions.
We recently reported in detail the cascade addition–cyclization–trapping reaction of substrates with carbon–carbon double bonds as two kinds of polarity-different radical acceptors [42]. On the basis of these results, the possibility of the carbon–carbon triple bond as a radical acceptor and the hydroxamate ester functionality as a two-point-binding coordination tether was next studied in detail. To understand the scope and limitation of the cascade transformation of hydroxamate esters with carbon–carbon triple bonds, the substrates of choice were 5, 6A–C, 7 and 8 having hydroxamate ester functionality (Figure 3).
![[1860-5397-9-128-3]](/bjoc/content/figures/1860-5397-9-128-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Substrates for testing the cascade transformation.
Figure 3: Substrates for testing the cascade transformation.
At first, we studied the cascade reaction of 5 with an acryloyl moiety and 6A–C with a methacryloyl moiety as an electron-deficient acceptor in the absence of a chiral ligand (Scheme 2). To control the rotamer population of substrates, Zn(OTf)2 was used as a Lewis acid to coordinate the hydroxamate ester functionality. The reactions were evaluated in CH2Cl2 at 20 °C under the tin-free iodine atom transfer conditions by using isopropyl iodide and Et3B. The reaction of hydroxamate ester 5 did not give the desired product probably due to polymerization of 5 through the labile acrylamide moiety. In contrast, the reaction of 6A–C proceeded effectively to give the cyclic products 9Aa–9Ca in good yields. Among them, hydroxamate esters 6A and 6B, which have a small methyl or benzyl group as R1, have shown a high reactivity, although a 76% yield of product 9Ca was obtained even when hydroxamate ester 6C having a 2-naphthylmethyl group was used. Furthermore, the regiochemical course of the initial radical addition to 6A–C was well controlled. The nucleophilic isopropyl radical reacted selectively with the electron-deficient methacryloyl moiety to give the single isomers 9Aa–9Ca.
![[1860-5397-9-128-i2]](/bjoc/content/inline/1860-5397-9-128-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cascade radical addition–cyclization–trapping reaction of 5 and 6A–C.
Scheme 2: Cascade radical addition–cyclization–trapping reaction of 5 and 6A–C.
It is also important to note that Z-isomers 9Aa–9Ca were selectively obtained without the formation of corresponding E-isomers. The E,Z-selectivities are determined by capturing the intermediate vinyl radicals with an atom-transfer reagent such as isopropyl iodide (Figure 4). These selectivities are controlled by the steric factor around vinyl radicals. The vinyl radicals are σ-radicals in a very fast equilibrium between E-isomer B and Z-isomer C. The steric hindrance between the substituents on the α-carbon atom of radical C and isopropyl iodide is assumed to lead to selective iodine atom-transfer in radical B giving 9Aa–9Ca as single Z-isomers.
![[1860-5397-9-128-4]](/bjoc/content/figures/1860-5397-9-128-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
On the basis of the above results, we next studied the reaction of 6A–C at −78 °C in the presence of Zn(OTf)2 and chiral box ligands L1–L3 (Scheme 3 and Table 1). A stoichiometric amount of chiral Lewis acid prepared from Zn(OTf)2 and ligand L1 accelerated the reaction of hydroxamate ester 6A having a methyl group as substituent R1 (Table 1, entry 1), although the reaction of 6A did not proceed effectively at −78 °C in the absence of box ligand L1. The desired product 9Aa was isolated as a single isomer in 51% yield with 60% ee after being stirred for 10 h. The use of hydroxamate ester 6B having a benzyl group led to not only an enhancement in chemical yield but also to an improvement in enantioselectivity to give the product 9Ba in 87% yield with 80% ee (Table 1, entry 2). Next, the catalytic nature of the reactions was examined (Table 1, entries 3–5). The reactions proceeded equally well with 50 and 30 mol % of chiral Lewis acid as with a stoichiometric amount (Table 1, entry 3 and 4). Further reduction of the chiral Lewis acid load to 10 mol % resulted in a decrease of both the chemical yield and enantioselectivity (Table 1, entry 5). In the case of 10 mol % of the chiral Lewis acid, the ternary complex of the ligand, the Lewis acid and the substrate were not effectively formed, and the background reaction giving the racemic product proceeded. Additionally, the high Z-selectivity of product 9Ba indicates that the stereoselective iodine-atom transfer from isopropyl iodide to an intermediate radical proceeded effectively under these catalytic reaction conditions. The reaction using box ligand L2 instead of L1 attenuated the enantioselectivity (Table 1, entry 6). A somewhat lower enantioselectivity was obtained by using ligand L3, surprisingly resulting in antipode product 9Ba (Table 1, entry 7). The representative effect of the solvent is shown in Table 1, entries 8–10. No reaction occurred in toluene, owing to the low solubility of the chiral Lewis acid in toluene (Table 1, entry 8). When the reaction was carried out in toluene/CH2Cl2 (4:1, v/v), the cyclic product 9Ba was obtained in 67% yield with 77% ee (Table 1, entry 9). The reaction in the protic solvent MeOH gave the nearly racemic product, although the high Z-selectivity was maintained (Table 1, entry 10). These results suggest that the rigid chelation of the chiral Lewis acid to the hydroxamate ester functionality occured in CH2Cl2. In the presence of chiral Lewis acid, hydroxamate ester 6C had also shown good reactivity, although the enantioselectivity diminished to 75% ee (Table 1, entry 11). We next studied the reaction of substrate 6B with other radical precursors (Table 1, entries 12–14). Reactions with cyclohexyl and cyclopentyl radicals were also facile. Under analogous reaction conditions, an outstanding level of enantioselectivity was observed on employing the bulky tert-butyl iodide as a radical precursor (Table 1, entry 14). A good yield of the product 9Bd was obtained with 92% ee and high Z-selectivity.
![[1860-5397-9-128-i3]](/bjoc/content/inline/1860-5397-9-128-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Enantioselective cascade reaction of 6A–C.
Scheme 3: Enantioselective cascade reaction of 6A–C.
Table 1: Reaction of 6A–C in the presence of chiral ligand.
| entry | substrate | R2 | ligand |
Lewis acid
(equiv) |
product
(% yield) |
Z/E | ee (%) |
|---|---|---|---|---|---|---|---|
| 1 | 6A | iPr | L1 | 1.0 | 9Aa (51) | >98:2 | 60 |
| 2 [39] | 6B | iPr | L1 | 1.0 | 9Ba (87) | >98:2 | 80 |
| 3 [39] | 6B | iPr | L1 | 0.5 | 9Ba (85) | >98:2 | 81 |
| 4 [39] | 6B | iPr | L1 | 0.3 | 9Ba (82) | >98:2 | 81 |
| 5 [39] | 6B | iPr | L1 | 0.1 | 9Ba (49)a | >98:2 | 47 |
| 6 | 6B | iPr | L2 | 1.0 | 9Ba (76) | >98:2 | 71 |
| 7 | 6B | iPr | L3 | 1.0 | 9Ba (81) | >98:2 | –69 |
| 8b | 6B | iPr | L1 | 1.0 | no reaction | ||
| 9c | 6B | iPr | L1 | 1.0 | 9Ba (67) | >98:2 | 77 |
| 10d | 6B | iPr | L1 | 1.0 | 9Ba (63) | >98:2 | rac |
| 11 | 6C | iPr | L1 | 1.0 | 9Ca (83) | >98:2 | 75 |
| 12 [39] | 6B | c-Hex | L1 | 1.0 | 9Bb (82) | >98:2 | 81 |
| 13 | 6B | c-Pent | L1 | 1.0 | 9Bc (83) | >98:2 | 79 |
| 14 [39] | 6B | t-Bu | L1 | 1.0 | 9Bd (85) | >98:2 | 92 |
astarting substrate 6B was recovered in 29% yield; bin toluene; cin toluene/CH2Cl2 (4:1, v/v); din MeOH.
The absolute configuration at the newly generated stereocenters of 9Aa–Bd was assumed by similarity between the present reaction and the previously reported reaction of substrates having the carbon–carbon double bond [39,42]. In these reactions, a ternary complex of ligand, Lewis acid and substrate would control the three-dimensional arrangement of two radical acceptors. A tetrahedral or cis-octahedral geometry around the zinc center was proposed [71,72]. In Figure 5, a tentative model of an octahedral complex is shown, in which two oxygen atoms of the hydroxamate ester functionality occupy two equatorial positions.
![[1860-5397-9-128-5]](/bjoc/content/figures/1860-5397-9-128-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Model for the enantioselective reaction.
Figure 5: Model for the enantioselective reaction.
To study the effect of an electron-deficient acceptor on the cascade process, the reactions of propiolic acid derivatives 7 and 8 were tested (Scheme 4). At first, the reaction of 7 was evaluated under asymmetric reaction conditions. However, the cascade addition–cyclization–trapping reaction did not proceed, and the simple adduct 10 was formed in 57% yield by the addition–trapping process. Next, the reaction of propiolic acid derivative 8 was tested, because we expected the [1,5]-hydrogen shift from 1,3-dioxolane ring into the reactive vinyl radical as shown as D. However, the simple adduct 11 was only obtained in 78% yield. The results from these studies show that a carbon–carbon double bond, e.g., a methacryloyl group, of the electron-deficient acceptor is essential for the successful cascade transformation.
![[1860-5397-9-128-i4]](/bjoc/content/inline/1860-5397-9-128-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of propiolic acid derivatives 7 and 8.
Scheme 4: Reaction of propiolic acid derivatives 7 and 8.
To gain further insight into the stereocontrol in the cyclization step, we next studied the opposite regiochemical cyclization by using the substrate 12 via the intermediate radical F (Scheme 5). The reaction was carried out in the presence of Bu3SnH under asymmetric reaction conditions. Although the reaction proceeded even at −78 °C, the nearly racemic product 13 was isolated in 60% yield. This observation indicates that the regiochemical course of the cyclization step is an important factor to achieve the highly asymmetric induction.
![[1860-5397-9-128-i5]](/bjoc/content/inline/1860-5397-9-128-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Opposite regiochemical cyclization using substrate 12.
Scheme 5: Opposite regiochemical cyclization using substrate 12.
We next investigated the reactivity of internal alkynes as electron-rich acceptors (Scheme 6). The internal alkyne 14 has shown a good reactivity comparable to that of the terminal alkynes 6A–C. In the absence of a chiral ligand, the zinc Lewis acid accelerated the reaction of alkyne 14 with an isopropyl radical at 20 °C to give the desired cyclic product 15a in 73% yield. Under analogous reaction conditions, both cyclohexyl iodide and cyclopentyl iodide worked well to give 15b and 15c in 65% and 68% yields, respectively. However, the reaction with a bulky tert-butyl radical did not proceed effectively, probably due to side reactions such as polymerization.
![[1860-5397-9-128-i6]](/bjoc/content/inline/1860-5397-9-128-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
We finally investigated the enantioselective reaction of internal alkynes 14 and 16 (Scheme 7). The reaction of 14 proceeded with good enantioselectivities (Table 2). When a stoichiometric amount of chiral Lewis acid was employed, the reaction with an isopropyl radical gave the desired product 15a in 86% yield with 83% ee (Table 2, entry 1). The reaction proceeded equally well with 30 mol % of chiral Lewis acid as with a stoichiometric amount (Table 2, entry 2). The secondary radicals, generated from cyclohexyl iodide or cyclopentyl iodide, reacted well to afford 15b and 15c with 85% ee and 83% ee, respectively (Table 2, entry 3 and 4). In marked contrast to the reaction in the absence of a chiral ligand (Scheme 6), the use of bulky tert-butyl iodide led to not only an enhancement in chemical yield but also to an improvement in enantioselectivity (Table 2, entry 5). These observations indicate that the combination of chiral Lewis acid and hydroxamate ester functionality led the rigid complex promoting the cyclization step and suppressing the background reaction or the undesired side reactions. High chemical yield and enantioselectivity were observed with 50 mol % of chiral Lewis acid (Table 2, entry 6), although further reduction of the catalyst load to 30 mol % resulted in a decrease of yield and enantioselectivity (Table 2, entry 7). Both chemical yield and enantioselectivity decreased by changing Lewis acid from Zn(OTf)2 to MgI2 (Table 2, entry 8). When the more nucleophilic and stable tert-butyl radical was employed, the reaction of substrate 16 having a phenyl group at the terminal position proceeded smoothly to give the desired product 17 in 89% yield with 67% ee (Table 2, entry 9). It is also important to note that the high Z/E-selectivity of products was observed even when internal alkynes 14 and 16 were employed. These results indicate that the iodine atom-transfer from R2I to the substituted vinyl radicals proceeded stereoselectively. Particularly, the substrate 16 having a phenyl group gave the intermediate linear π-radical. Thus, the capture of linear vinyl radical with atom-transfer reagent would be influenced by the steric hindrance around the quaternary carbon atom [43].
![[1860-5397-9-128-i7]](/bjoc/content/inline/1860-5397-9-128-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Enantioselective cascade reaction of 14 and 16.
Scheme 7: Enantioselective cascade reaction of 14 and 16.
Table 2: Reaction of 14 and 16 in the presence of a chiral ligand.
| entry | substrate | R2 | Lewis acid (equiv) | yield (%) | ratio | ee (%) |
|---|---|---|---|---|---|---|
| 1 [39] | 14 | iPr | Zn(OTf)2 (1.0) | 86 | >98:2 | 83 |
| 2 [39] | 14 | iPr | Zn(OTf)2 (0.3) | 74 | >98:2 | 81 |
| 3 [39] | 14 | c-Hex | Zn(OTf)2 (1.0) | 87 | >98:2 | 85 |
| 4 | 14 | c-Pent | Zn(OTf)2 (1.0) | 77 | >98:2 | 83 |
| 5 [39] | 14 | t-Bu | Zn(OTf)2 (1.0) | 94 | >98:2 | 90 |
| 6 | 14 | t-Bu | Zn(OTf)2 (0.5) | 94 | >98:2 | 91 |
| 7 | 14 | t-Bu | Zn(OTf)2 (0.3) | 75 | >98:2 | 61 |
| 8 | 14 | t-Bu | MgI2 (1.0) | 20 | >98:2 | 54 |
| 9 | 16 | t-Bu | Zn(OTf)2 (1.0) | 89 | >98:2 | 67 |
Conclusion
We have shown the cascade radical addition–cyclization–trapping reaction of substrates with a carbon–carbon triple bond as a radical acceptor as well as the scope and limitation of hydroxamate ester as a coordination site with a chiral Lewis acid. Synthetic strategies involving enantioselective radical cyclizations would be desirable tools for preparing functionalized cyclic compounds with multiple stereocenters. These studies offer opportunities for further exploration of fascinating possibilities in the realm of cascade radical reactions.
Supporting Information
| Supporting Information File 1: General experimental procedures, characterization data of obtained compounds, and preparation of substrates. | ||
| Format: PDF | Size: 663.4 KB | Download |
References
-
Smadja, W. Synlett 1994, 1–26. doi:10.1055/s-1994-22728
Return to citation in text: [1] -
Fallis, A. G.; Brinza, I. M. Tetrahedron 1997, 53, 17543–17594. doi:10.1016/S0040-4020(97)10060-6
Return to citation in text: [1] -
Renaud, P.; Gerster, M. Angew. Chem., Int. Ed. 1998, 37, 2562–2579. doi:10.1002/(SICI)1521-3773(19981016)37:19<2562::AID-ANIE2562>3.0.CO;2-D
Return to citation in text: [1] -
Sibi, M. P.; Porter, N. A. Acc. Chem. Res. 1999, 32, 163–171. doi:10.1021/ar9600547
Return to citation in text: [1] -
Naito, T. Heterocycles 1999, 50, 505–541. doi:10.3987/REV-98-SR(H)2
Return to citation in text: [1] -
Renaud, P.; Sibi, M. P., Eds. Radicals in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1 and 2.
Return to citation in text: [1] -
Bar, G.; Parsons, A. F. Chem. Soc. Rev. 2003, 32, 251–263. doi:10.1039/b111414j
Return to citation in text: [1] -
Sibi, M. P.; Manyem, S.; Zimmerman, J. Chem. Rev. 2003, 103, 3263–3296. doi:10.1021/cr020044l
Return to citation in text: [1] -
Srikanth, G. S. C.; Castle, S. L. Tetrahedron 2005, 61, 10377–10441. doi:10.1016/j.tet.2005.07.077
Return to citation in text: [1] -
Guo, H.-C.; Ma, J.-A. Angew. Chem., Int. Ed. 2006, 45, 354–366. doi:10.1002/anie.200500195
Return to citation in text: [1] -
Zimmerman, J.; Sibi, M. P. Top. Curr. Chem. 2006, 263, 107–162. doi:10.1007/128_027
Return to citation in text: [1] -
Godineau, E.; Landais, Y. Chem.–Eur. J. 2009, 15, 3044–3055. doi:10.1002/chem.200802415
Return to citation in text: [1] -
Rowlands, G. J. Tetrahedron 2009, 65, 8603–8655. doi:10.1016/j.tet.2009.07.001
Return to citation in text: [1] -
Rowlands, G. J. Tetrahedron 2010, 66, 1593–1636. doi:10.1016/j.tet.2009.12.023
Return to citation in text: [1] -
Perchyonok, V. T. Radical Reactions in Aqueous Media. In RSC Green Chemistry, No. 6, RSC Publishing: Cambridge, 2010.
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Yang, Y.-H.; Sibi, P. Stereoselective Radical Reactions. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; Wiley: Weinheim, Germany, 2012; Vol. 2, pp 655–692. doi:10.1002/9781119953678.rad019
Return to citation in text: [1] -
Ischay, M. A.; Yoon, T. P. Eur. J. Org. Chem. 2012, 3359–3372. doi:10.1002/ejoc.201101071
Return to citation in text: [1] -
Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f
Return to citation in text: [1] -
Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x
Return to citation in text: [1] -
Nozaki, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1988, 29, 1041–1044. doi:10.1016/0040-4039(88)85330-9
Return to citation in text: [1] -
Curran, D. P.; Chen, M. H.; Spletzer, E.; Seong, C. M.; Chang, C. T. J. Am. Chem. Soc. 1989, 111, 8872–8878. doi:10.1021/ja00206a016
Return to citation in text: [1] -
Giese, B.; Zehnder, M.; Roth, M.; Zeitz, H.-G. J. Am. Chem. Soc. 1990, 112, 6741–6742. doi:10.1021/ja00174a061
Return to citation in text: [1] -
Porter, N. A.; Rosenstein, I. J.; Breyer, R. A.; Bruhnke, J. D.; Wu, W. X.; McPhail, A. T. J. Am. Chem. Soc. 1992, 114, 7664–7676. doi:10.1021/ja00046a010
Return to citation in text: [1] -
Marco-Contelles, J. Chem. Commun. 1996, 2629–2630. doi:10.1039/cc9960002629
Return to citation in text: [1] -
Takai, K.; Matsukawa, N.; Takahashi, A.; Fujii, T. Angew. Chem., Int. Ed. 1998, 37, 152–155. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<152::AID-ANIE152>3.3.CO;2-#
Return to citation in text: [1] -
Miyabe, H.; Fujii, K.; Goto, T.; Naito, T. Org. Lett. 2000, 2, 4071–4074. doi:10.1021/ol006716g
Return to citation in text: [1] -
Sibi, M. P.; Chen, J. J. Am. Chem. Soc. 2001, 123, 9472–9473. doi:10.1021/ja016633a
Return to citation in text: [1] -
Dhimane, A.-L.; Aïssa, C.; Malacria, M. Angew. Chem., Int. Ed. 2002, 41, 3284–3287. doi:10.1002/1521-3773(20020902)41:17<3284::AID-ANIE3284>3.0.CO;2-Z
Return to citation in text: [1] -
Yamago, S.; Miyoshi, M.; Miyazoe, H.; Yoshida, J. Angew. Chem., Int. Ed. 2002, 41, 1407–1409. doi:10.1002/1521-3773(20020415)41:8<1407::AID-ANIE1407>3.0.CO;2-Z
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e
Return to citation in text: [1] -
Tsuchii, K.; Doi, M.; Hirao, T.; Ogawa, A. Angew. Chem., Int. Ed. 2003, 42, 3490–3493. doi:10.1002/anie.200250790
Return to citation in text: [1] -
Denes, F.; Chemla, F.; Normant, J. F. Angew. Chem., Int. Ed. 2003, 42, 4043–4046. doi:10.1002/anie.200250474
Return to citation in text: [1] -
Yamamoto, Y.; Nakano, S.; Maekawa, H.; Nishiguchi, I. Org. Lett. 2004, 6, 799–802. doi:10.1021/ol036506e
Return to citation in text: [1] -
Uenoyama, Y.; Fukuyama, T.; Nobuta, O.; Matsubara, H.; Ryu, I. Angew. Chem., Int. Ed. 2005, 44, 1075–1078. doi:10.1002/anie.200461954
Return to citation in text: [1] -
Ueda, U.; Miyabe, H.; Sugino, H.; Miyata, O.; Naito, T. Angew. Chem., Int. Ed. 2005, 44, 6190–6193. doi:10.1002/anie.200502263
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Yoshida, K.; Takemoto, Y. Synlett 2004, 540–542. doi:10.1055/s-2004-815407
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron 2005, 61, 385–393. doi:10.1016/j.tet.2004.10.104
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] -
Miyabe, H.; Toyoda, A.; Takemoto, Y. Synlett 2007, 1885–1888. doi:10.1055/s-2007-984530
Return to citation in text: [1] [2] -
Yoshioka, E.; Kentefu; Wang, X.; Kohtani, S.; Miyabe, H. Synlett 2011, 2085–2089. doi:10.1055/s-0030-1261167
Return to citation in text: [1] [2] -
Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Yoshioka, E.; Kohtani, S.; Sawai, K.; Kentefu; Tanaka, E.; Miyabe, H. J. Org. Chem. 2012, 77, 8588–8604. doi:10.1021/jo3015227
Return to citation in text: [1] [2] [3] -
Nishida, M.; Hayashi, H.; Nishida, A.; Kawahara, N. Chem. Commun. 1996, 579–580. doi:10.1039/cc9960000579
Return to citation in text: [1] -
Hiroi, K.; Ishii, M. Tetrahedron Lett. 2000, 41, 7071–7074. doi:10.1016/S0040-4039(00)01213-2
Return to citation in text: [1] -
Yang, D.; Gu, S.; Yan, Y.-L.; Zhu, N.-Y.; Cheung, K.-K. J. Am. Chem. Soc. 2001, 123, 8612–8613. doi:10.1021/ja016383y
Return to citation in text: [1] -
Yang, D.; Gu, S.; Yan, Y.-L.; Zhao, H.-W.; Zhu, N.-Y. Angew. Chem., Int. Ed. 2002, 41, 3014–3017. doi:10.1002/1521-3773(20020816)41:16<3014::AID-ANIE3014>3.0.CO;2-J
Return to citation in text: [1] -
Yang, D.; Zheng, B.-F.; Gao, Q.; Gu, S.; Zhu, N.-Y. Angew. Chem., Int. Ed. 2006, 45, 255–258. doi:10.1002/anie.200503056
Return to citation in text: [1] -
Aechtner, T.; Dressel, M.; Bach, T. Angew. Chem., Int. Ed. 2004, 43, 5849–5851. doi:10.1002/anie.200461222
Return to citation in text: [1] -
Bauer, A.; Westkämper, F.; Grimme, S.; Bach, T. Nature 2005, 436, 1139–1140. doi:10.1038/nature03955
Return to citation in text: [1] -
Breitenlechner, S.; Bach, T. Angew. Chem., Int. Ed. 2008, 47, 7957–7959. doi:10.1002/anie.200802479
Return to citation in text: [1] -
Beeson, T. D.; Mastracchio, A.; Hong, J.-B.; Ashton, K.; MacMillan, D. W. C. Science 2007, 316, 582–585.
Return to citation in text: [1] -
Jang, H.-Y.; Hong, J.-B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2007, 129, 7004–7005. doi:10.1021/ja0719428
Return to citation in text: [1] -
Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902
Return to citation in text: [1] -
Jui, N. T.; Lee, E. C. Y.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 10015–10017. doi:10.1021/ja104313x
Return to citation in text: [1] -
Rendler, S.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 5027–5029. doi:10.1021/ja100185p
Return to citation in text: [1] -
Pham, P. V.; Ashton, K.; MacMillan, D. W. C. Chem. Sci. 2011, 2, 1470–1473. doi:10.1039/c1sc00176k
Return to citation in text: [1] -
Jui, N. T.; Garber, J. A. O.; Finelli, F. G.; MacMillan, D. W. C. J. Am. Chem. Soc. 2012, 134, 11400–11403. doi:10.1021/ja305076b
Return to citation in text: [1] -
Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 2086–2087. doi:10.1021/ja809405c
Return to citation in text: [1] -
Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 6640. doi:10.1021/ja902816z
Return to citation in text: [1] -
Gansäuer, A.; Shi, L.; Otte, M. J. Am. Chem. Soc. 2010, 132, 11858–11859. doi:10.1021/ja105023y
Return to citation in text: [1] -
Gansäuer, A.; Behlendorf, M.; von Laufenberg, D.; Fleckhaus, A.; Kube, C.; Sadasivam, D. V.; Flowers, R. A., II. Angew. Chem., Int. Ed. 2012, 51, 4739–4742. doi:10.1002/anie.201200431
Return to citation in text: [1] -
Gansäuer, A.; Klatte, M.; Brändle, G. M.; Friedrich, J. Angew. Chem., Int. Ed. 2012, 51, 8891–8894. doi:10.1002/anie.201202818
Return to citation in text: [1] -
Streuff, J.; Feurer, M.; Bichovski, P.; Frey, G.; Gellrich, U. Angew. Chem., Int. Ed. 2012, 51, 8661–8664. doi:10.1002/anie.201204469
Return to citation in text: [1] -
Curran, D. P.; Liu, W.; Chen, C. H.-T. J. Am. Chem. Soc. 1999, 121, 11012–11013. doi:10.1021/ja993329x
Return to citation in text: [1] -
Nechab, M.; Campolo, D.; Maury, J.; Perfetti, P.; Vanthuyne, N.; Siri, D.; Bertrand, M. P. J. Am. Chem. Soc. 2010, 132, 14742–14744. doi:10.1021/ja106668d
Return to citation in text: [1] -
Mondal, S.; Nechab, M.; Campolo, D.; Vanthuyne, N.; Bertrand, M. P. Adv. Synth. Catal. 2012, 354, 1987–2000. doi:10.1002/adsc.201200045
Return to citation in text: [1] -
Mondal, S.; Nechab, M.; Vanthuyne, N.; Bertrand, M. P. Chem. Commun. 2012, 48, 2549–2551. doi:10.1039/c2cc17830c
Return to citation in text: [1] -
Corminboeuf, O.; Renaud, P. Org. Lett. 2002, 4, 1731–1734. doi:10.1021/ol025799t
Return to citation in text: [1] -
Corminboeuf, O.; Renaud, P. Org. Lett. 2002, 4, 1735–1738. doi:10.1021/ol0257981
Return to citation in text: [1] -
Sibi, M. P.; Yang, Y.-H. Synlett 2008, 83–88. doi:10.1055/s-2007-992386
Return to citation in text: [1] -
Evans, D. A.; Kozlowski, M. C.; Tedrow, J. S. Tetrahedron Lett. 1996, 37, 7481–7484. doi:10.1016/0040-4039(96)01697-8
Return to citation in text: [1]
| 43. | Yoshioka, E.; Kohtani, S.; Sawai, K.; Kentefu; Tanaka, E.; Miyabe, H. J. Org. Chem. 2012, 77, 8588–8604. doi:10.1021/jo3015227 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 42. | Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j |
| 71. | Sibi, M. P.; Yang, Y.-H. Synlett 2008, 83–88. doi:10.1055/s-2007-992386 |
| 72. | Evans, D. A.; Kozlowski, M. C.; Tedrow, J. S. Tetrahedron Lett. 1996, 37, 7481–7484. doi:10.1016/0040-4039(96)01697-8 |
| 1. | Smadja, W. Synlett 1994, 1–26. doi:10.1055/s-1994-22728 |
| 2. | Fallis, A. G.; Brinza, I. M. Tetrahedron 1997, 53, 17543–17594. doi:10.1016/S0040-4020(97)10060-6 |
| 3. | Renaud, P.; Gerster, M. Angew. Chem., Int. Ed. 1998, 37, 2562–2579. doi:10.1002/(SICI)1521-3773(19981016)37:19<2562::AID-ANIE2562>3.0.CO;2-D |
| 4. | Sibi, M. P.; Porter, N. A. Acc. Chem. Res. 1999, 32, 163–171. doi:10.1021/ar9600547 |
| 5. | Naito, T. Heterocycles 1999, 50, 505–541. doi:10.3987/REV-98-SR(H)2 |
| 6. | Renaud, P.; Sibi, M. P., Eds. Radicals in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1 and 2. |
| 7. | Bar, G.; Parsons, A. F. Chem. Soc. Rev. 2003, 32, 251–263. doi:10.1039/b111414j |
| 8. | Sibi, M. P.; Manyem, S.; Zimmerman, J. Chem. Rev. 2003, 103, 3263–3296. doi:10.1021/cr020044l |
| 9. | Srikanth, G. S. C.; Castle, S. L. Tetrahedron 2005, 61, 10377–10441. doi:10.1016/j.tet.2005.07.077 |
| 10. | Guo, H.-C.; Ma, J.-A. Angew. Chem., Int. Ed. 2006, 45, 354–366. doi:10.1002/anie.200500195 |
| 11. | Zimmerman, J.; Sibi, M. P. Top. Curr. Chem. 2006, 263, 107–162. doi:10.1007/128_027 |
| 12. | Godineau, E.; Landais, Y. Chem.–Eur. J. 2009, 15, 3044–3055. doi:10.1002/chem.200802415 |
| 13. | Rowlands, G. J. Tetrahedron 2009, 65, 8603–8655. doi:10.1016/j.tet.2009.07.001 |
| 14. | Rowlands, G. J. Tetrahedron 2010, 66, 1593–1636. doi:10.1016/j.tet.2009.12.023 |
| 15. | Perchyonok, V. T. Radical Reactions in Aqueous Media. In RSC Green Chemistry, No. 6, RSC Publishing: Cambridge, 2010. |
| 16. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 17. | Yang, Y.-H.; Sibi, P. Stereoselective Radical Reactions. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C.; Studer, A., Eds.; Wiley: Weinheim, Germany, 2012; Vol. 2, pp 655–692. doi:10.1002/9781119953678.rad019 |
| 18. | Ischay, M. A.; Yoon, T. P. Eur. J. Org. Chem. 2012, 3359–3372. doi:10.1002/ejoc.201101071 |
| 19. | Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f |
| 20. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 40. | Miyabe, H.; Toyoda, A.; Takemoto, Y. Synlett 2007, 1885–1888. doi:10.1055/s-2007-984530 |
| 41. | Yoshioka, E.; Kentefu; Wang, X.; Kohtani, S.; Miyabe, H. Synlett 2011, 2085–2089. doi:10.1055/s-0030-1261167 |
| 42. | Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j |
| 43. | Yoshioka, E.; Kohtani, S.; Sawai, K.; Kentefu; Tanaka, E.; Miyabe, H. J. Org. Chem. 2012, 77, 8588–8604. doi:10.1021/jo3015227 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 44. | Nishida, M.; Hayashi, H.; Nishida, A.; Kawahara, N. Chem. Commun. 1996, 579–580. doi:10.1039/cc9960000579 |
| 45. | Hiroi, K.; Ishii, M. Tetrahedron Lett. 2000, 41, 7071–7074. doi:10.1016/S0040-4039(00)01213-2 |
| 46. | Yang, D.; Gu, S.; Yan, Y.-L.; Zhu, N.-Y.; Cheung, K.-K. J. Am. Chem. Soc. 2001, 123, 8612–8613. doi:10.1021/ja016383y |
| 47. | Yang, D.; Gu, S.; Yan, Y.-L.; Zhao, H.-W.; Zhu, N.-Y. Angew. Chem., Int. Ed. 2002, 41, 3014–3017. doi:10.1002/1521-3773(20020816)41:16<3014::AID-ANIE3014>3.0.CO;2-J |
| 48. | Yang, D.; Zheng, B.-F.; Gao, Q.; Gu, S.; Zhu, N.-Y. Angew. Chem., Int. Ed. 2006, 45, 255–258. doi:10.1002/anie.200503056 |
| 49. | Aechtner, T.; Dressel, M.; Bach, T. Angew. Chem., Int. Ed. 2004, 43, 5849–5851. doi:10.1002/anie.200461222 |
| 50. | Bauer, A.; Westkämper, F.; Grimme, S.; Bach, T. Nature 2005, 436, 1139–1140. doi:10.1038/nature03955 |
| 51. | Breitenlechner, S.; Bach, T. Angew. Chem., Int. Ed. 2008, 47, 7957–7959. doi:10.1002/anie.200802479 |
| 52. | Beeson, T. D.; Mastracchio, A.; Hong, J.-B.; Ashton, K.; MacMillan, D. W. C. Science 2007, 316, 582–585. |
| 53. | Jang, H.-Y.; Hong, J.-B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2007, 129, 7004–7005. doi:10.1021/ja0719428 |
| 54. | Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902 |
| 55. | Jui, N. T.; Lee, E. C. Y.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 10015–10017. doi:10.1021/ja104313x |
| 56. | Rendler, S.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 5027–5029. doi:10.1021/ja100185p |
| 57. | Pham, P. V.; Ashton, K.; MacMillan, D. W. C. Chem. Sci. 2011, 2, 1470–1473. doi:10.1039/c1sc00176k |
| 58. | Jui, N. T.; Garber, J. A. O.; Finelli, F. G.; MacMillan, D. W. C. J. Am. Chem. Soc. 2012, 134, 11400–11403. doi:10.1021/ja305076b |
| 59. | Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 2086–2087. doi:10.1021/ja809405c |
| 60. | Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 6640. doi:10.1021/ja902816z |
| 61. | Gansäuer, A.; Shi, L.; Otte, M. J. Am. Chem. Soc. 2010, 132, 11858–11859. doi:10.1021/ja105023y |
| 62. | Gansäuer, A.; Behlendorf, M.; von Laufenberg, D.; Fleckhaus, A.; Kube, C.; Sadasivam, D. V.; Flowers, R. A., II. Angew. Chem., Int. Ed. 2012, 51, 4739–4742. doi:10.1002/anie.201200431 |
| 63. | Gansäuer, A.; Klatte, M.; Brändle, G. M.; Friedrich, J. Angew. Chem., Int. Ed. 2012, 51, 8891–8894. doi:10.1002/anie.201202818 |
| 64. | Streuff, J.; Feurer, M.; Bichovski, P.; Frey, G.; Gellrich, U. Angew. Chem., Int. Ed. 2012, 51, 8661–8664. doi:10.1002/anie.201204469 |
| 65. | Curran, D. P.; Liu, W.; Chen, C. H.-T. J. Am. Chem. Soc. 1999, 121, 11012–11013. doi:10.1021/ja993329x |
| 66. | Nechab, M.; Campolo, D.; Maury, J.; Perfetti, P.; Vanthuyne, N.; Siri, D.; Bertrand, M. P. J. Am. Chem. Soc. 2010, 132, 14742–14744. doi:10.1021/ja106668d |
| 67. | Mondal, S.; Nechab, M.; Campolo, D.; Vanthuyne, N.; Bertrand, M. P. Adv. Synth. Catal. 2012, 354, 1987–2000. doi:10.1002/adsc.201200045 |
| 68. | Mondal, S.; Nechab, M.; Vanthuyne, N.; Bertrand, M. P. Chem. Commun. 2012, 48, 2549–2551. doi:10.1039/c2cc17830c |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 37. | Miyabe, H.; Asada, R.; Yoshida, K.; Takemoto, Y. Synlett 2004, 540–542. doi:10.1055/s-2004-815407 |
| 38. | Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron 2005, 61, 385–393. doi:10.1016/j.tet.2004.10.104 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 40. | Miyabe, H.; Toyoda, A.; Takemoto, Y. Synlett 2007, 1885–1888. doi:10.1055/s-2007-984530 |
| 41. | Yoshioka, E.; Kentefu; Wang, X.; Kohtani, S.; Miyabe, H. Synlett 2011, 2085–2089. doi:10.1055/s-0030-1261167 |
| 42. | Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j |
| 43. | Yoshioka, E.; Kohtani, S.; Sawai, K.; Kentefu; Tanaka, E.; Miyabe, H. J. Org. Chem. 2012, 77, 8588–8604. doi:10.1021/jo3015227 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 21. | Nozaki, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1988, 29, 1041–1044. doi:10.1016/0040-4039(88)85330-9 |
| 22. | Curran, D. P.; Chen, M. H.; Spletzer, E.; Seong, C. M.; Chang, C. T. J. Am. Chem. Soc. 1989, 111, 8872–8878. doi:10.1021/ja00206a016 |
| 23. | Giese, B.; Zehnder, M.; Roth, M.; Zeitz, H.-G. J. Am. Chem. Soc. 1990, 112, 6741–6742. doi:10.1021/ja00174a061 |
| 24. | Porter, N. A.; Rosenstein, I. J.; Breyer, R. A.; Bruhnke, J. D.; Wu, W. X.; McPhail, A. T. J. Am. Chem. Soc. 1992, 114, 7664–7676. doi:10.1021/ja00046a010 |
| 25. | Marco-Contelles, J. Chem. Commun. 1996, 2629–2630. doi:10.1039/cc9960002629 |
| 26. | Takai, K.; Matsukawa, N.; Takahashi, A.; Fujii, T. Angew. Chem., Int. Ed. 1998, 37, 152–155. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<152::AID-ANIE152>3.3.CO;2-# |
| 27. | Miyabe, H.; Fujii, K.; Goto, T.; Naito, T. Org. Lett. 2000, 2, 4071–4074. doi:10.1021/ol006716g |
| 28. | Sibi, M. P.; Chen, J. J. Am. Chem. Soc. 2001, 123, 9472–9473. doi:10.1021/ja016633a |
| 29. | Dhimane, A.-L.; Aïssa, C.; Malacria, M. Angew. Chem., Int. Ed. 2002, 41, 3284–3287. doi:10.1002/1521-3773(20020902)41:17<3284::AID-ANIE3284>3.0.CO;2-Z |
| 30. | Yamago, S.; Miyoshi, M.; Miyazoe, H.; Yoshida, J. Angew. Chem., Int. Ed. 2002, 41, 1407–1409. doi:10.1002/1521-3773(20020415)41:8<1407::AID-ANIE1407>3.0.CO;2-Z |
| 31. | Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e |
| 32. | Tsuchii, K.; Doi, M.; Hirao, T.; Ogawa, A. Angew. Chem., Int. Ed. 2003, 42, 3490–3493. doi:10.1002/anie.200250790 |
| 33. | Denes, F.; Chemla, F.; Normant, J. F. Angew. Chem., Int. Ed. 2003, 42, 4043–4046. doi:10.1002/anie.200250474 |
| 34. | Yamamoto, Y.; Nakano, S.; Maekawa, H.; Nishiguchi, I. Org. Lett. 2004, 6, 799–802. doi:10.1021/ol036506e |
| 35. | Uenoyama, Y.; Fukuyama, T.; Nobuta, O.; Matsubara, H.; Ryu, I. Angew. Chem., Int. Ed. 2005, 44, 1075–1078. doi:10.1002/anie.200461954 |
| 36. | Ueda, U.; Miyabe, H.; Sugino, H.; Miyata, O.; Naito, T. Angew. Chem., Int. Ed. 2005, 44, 6190–6193. doi:10.1002/anie.200502263 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 42. | Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 69. | Corminboeuf, O.; Renaud, P. Org. Lett. 2002, 4, 1731–1734. doi:10.1021/ol025799t |
| 70. | Corminboeuf, O.; Renaud, P. Org. Lett. 2002, 4, 1735–1738. doi:10.1021/ol0257981 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
| 42. | Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j |
| 42. | Miyabe, H.; Asada, R.; Takemoto, Y. Org. Biomol. Chem. 2012, 10, 3519–3530. doi:10.1039/c2ob25073j |
| 39. | Miyabe, H.; Asada, R.; Toyoda, A.; Takemoto, Y. Angew. Chem., Int. Ed. 2006, 45, 5863–5866. doi:10.1002/anie.200602042 |
© 2013 Miyabe et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)