Abstract
A successful enone version of an intramolecular aza-[3 + 3] annulation reaction is described here. Use of piperidinium trifluoroacetate salt as the catalyst and toluene as the solvent appears to be critical for a successful annulation. We also demonstrated for the first time that microwave irradiation can accelerate aza-[3 + 3] annulation reactions. An attempt to expand the scope of the enone aza-[3 + 3] annulation was made in the form of propyleine synthesis as a proof of concept. While synthesis of the enone annulation precursor was successfully accomplished, the annulation proved to be challenging and was only modestly successful.

Graphical Abstract
Introduction
Throughout the past decade, we have been developing an aza-[3 + 3] annulation reaction as a general and unified strategy in alkaloid synthesis [1-32]. Our aza-[3 + 3] annulation, which has been classified as Type-II [1], with Type-I aza-[3 + 3] annulation being reserved for Robinson’s double Mannich-type process [33], utilizes readily accessible and easily handled vinylogous amides and vinyl iminium salts. It provides a significant complementary, if not superior, approach to aza-[4 + 2] cycloadditions in constructing piperidines, because the aza-dienes and imines required are not always the most accessible and/or easily handled substrates given the problems of isomerization and hydrolysis (Figure 1) [34-37].
![[1860-5397-9-131-1]](/bjoc/content/figures/1860-5397-9-131-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
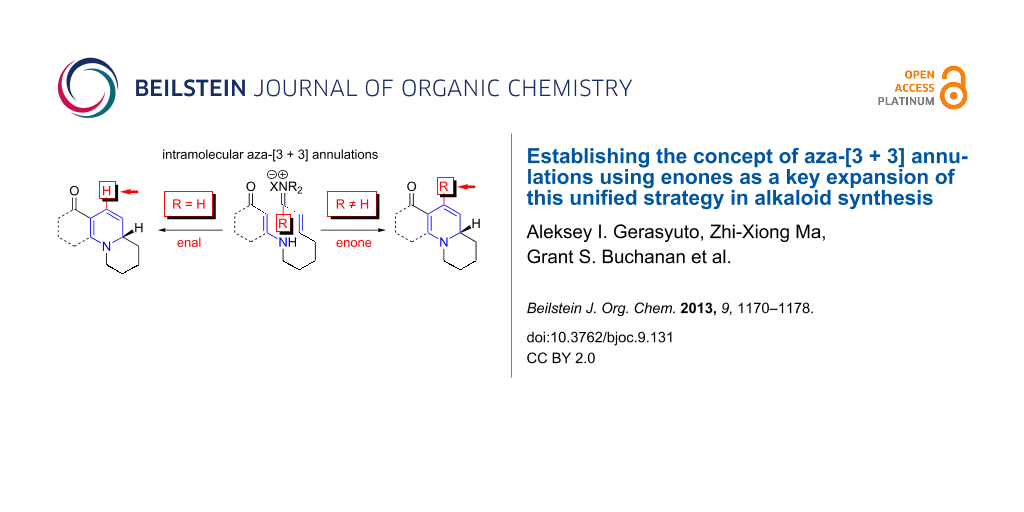
The prevalence of six-membered nitrogen heterocyclic motifs in alkaloids renders the development of this aza-[3 + 3] annulation into a powerful strategy a unique opportunity in the field of alkaloid synthesis [1-4,8-15]. The intramolecular variant of this annulation has proven to be particularly valuable for total synthesis [16,26-32]. Specifically, the intramolecular aza-[3 + 3] annulation of vinylogous amides tethered to a vinyl iminium motif 1a proceeds through a tandem sequence of N-1,4-addition and C-1,2-addition/β-elimination [16,17], effectively leading to a variety of nitrogen heterocyclic motifs, such as 3, which are prevalent among alkaloids (Scheme 1) [16,26-32]. However, there was a significant deficiency in this annulation: the inability to employ vinylogous amides tethered to α,β-unsaturated ketones, or enones 1b (R ≠ H) (Scheme 1).
![[1860-5397-9-131-i1]](/bjoc/content/inline/1860-5397-9-131-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Aza-[3 + 3] annulations with enones.
Scheme 1: Aza-[3 + 3] annulations with enones.
Conceptually, employing enals or enones in an aza-[3 + 3] annulation appears to constitute very little difference, with the major difference lying in the R group in their respective products 3 and 4. While seemingly an insignificant perturbation, the ability to employ enones in aza-[3 + 3] annulations can generate a vast array of opportunities, including the case where R is as small and simple as a Me group, which would already represent a facile entry to aza-phenylene alkaloids [30,31,38-45], such as propyleine [46-48], as well as lycopodium alkaloids that are rich in bioactivities [49-51] (Figure 2). In addition, the importance of these ventures into an aza-[3 + 3] annulation with enones can also be further demonstrated through syntheses of tetrahydroisoquinolines and indole alkaloids [52-55] in which the R group can constitute a larger functionality of the alkaloids, such as that seen in geissoschizine [52-57], and ipecac alkaloids such as protoemetine [48,52-55,58-61], emetine [52-55,58-63], and tubulosine [64]. With the appropriate choice of an R group in the synthetic design, a highly convergent and expedient strategy can come forth for constructing these alkaloids. We report herein our success in achieving intramolecular aza-[3 + 3] annulations of an enone.
![[1860-5397-9-131-2]](/bjoc/content/figures/1860-5397-9-131-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Possible natural-product targets.
Figure 2: Possible natural-product targets.
Results and Discussion
To examine the feasibility of an enone intramolecular aza-[3 + 3] annulation, a seven-step synthesis of the annulation substrate 10 commencing from 3-butyn-2-ol (5) was carried out (Scheme 2). Protection of secondary propargyl alcohol 5 as the THP-ether followed by alkylation of a lithium acetylide onto 1,4-dibromobutane afforded bromide 6 in 81% overall yield. Acid-mediated removal of THP followed by azide formation using NaN3 afforded alkyl azide 7 in 94% overall yield.
![[1860-5397-9-131-i2]](/bjoc/content/inline/1860-5397-9-131-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the annulation precursor enone 10.
Scheme 2: Synthesis of the annulation precursor enone 10.
Preparation of vinylogous amide 9 was then achieved by a two-step sequence: (a) LAH-mediated azide reduction to give a primary amine with concomitant reduction of the alkyne revealing the olefin functionality; and (b) dehydrative condensation of this primary amine with 1,3-cyclohexanedione. Oxidation of the secondary allylic alcohol in 9 with MnO2 at ambient temperature provided enone 10 in 85% yield. We were poised to investigate the viability of the enone aza-[3 + 3] annulation.
Our initial efforts to employ enone substrate 10 met with failure when using up to 500 mol % of piperidinium chloride [65] (Table 1, entry 1) or 100 mol % piperidinium acetate salts (Table 1, entry 2); even at higher temperatures of 120 °C, these chloride and acetate salts proved to be ineffective (Table 1, entry 3). We then explored salts prepared from stronger acids such as (+)-camphorsulfonic acid (CSA) and trifluoroacetic acid, which should tend to dissociate easier in the reaction medium and lead to a more reactive vinyl iminium salt intermediate 1b. We were elated to observe that the use of 100 mol % of piperidinium CSA salt in toluene at 120 °C provided 100% conversion with 60% yield of cycloadduct 11 (Table 1, entry 4). Further optimizations using trifluoroacetate salt (Table 1, entries 5–8) revealed that the yield could be improved to 87%, while lowering the catalyst loading from 500 mol % to 50 mol %, although the reaction temperature was raised to 150 °C. Lastly, we also examined the use of microwave irradiation [66,67] and observed a distinct rate enhancement (Table 1, entry 9).
Table 1: Catalyst screening and optimization.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-131-i6.svg?max-width=637&scale=1.0)
|
||||||
| entrya | HX (mol %) | solvent | temp (°C) | time (h) | conv. (%) | yield (%)b |
|---|---|---|---|---|---|---|
| 1 | HCl (500) | EtOAc/toluene | 100 | 16 | 0 | – |
| 2 | HOAc (100) | EtOAc | 85 | 12 | 0 | – |
| 3 | HOAc (500) | toluene | 120 | 16 | 0 | – |
| 4 | (+)-CSAc (100) | toluene | 120 | 12 | 100 | 60 |
| 5 | HO2CCF3 (500) | toluene | 85 | 7 | 79 | 39 |
| 6 | HO2CCF3 (100) | toluene | 85 | 5 | 100 | 50 |
| 7 | HO2CCF3 (100) | EtOAc/toluene | 110 | 4 | 92 | 41 |
| 8 | HO2CCF3 (50) | toluene | 150 | 3 | 100 | 87 |
| 9 | HO2CCF3 (90) | toluene | MWd | 1.5 | 94 | 65 |
aAll reactions run in a sealed tube. Concn = 0.03–0.04 M; 5.0 equiv Na2SO4. bIsolated yields. c(+)-Camphorusulfonic acid. d450-W Microwave.
The observed reactivity difference of the trifluoroacetate and camphorsulfonic acid salts, as compared to the chloride and acetate salts, can be attributed to the rate at which they promote vinyl iminium ion formation through a “balanced act” [12]. The difference in reactivity is related to the dissociation capacity of the respective amine salts. Since both “free amine” and “free acid” are needed in a synergistic manner to generate the vinyl iminium ion from the corresponding enone, the ability of the amine salt to dissociate to its “free amine” and “free acid” can exert an impact on the rate of the iminium formation. The low reactivity of the chloride salt can be attributed to its higher resistance toward dissociation, or is simply a tighter ion-pair compared to the CSA and the trifluoroacetate salt. At the same time, the formation of vinyl iminium ion from carbonyl systems and the “free amine” is also promoted by protonation of the carbonyl group via the “free acid”. Consequently, the increased reactivity of the CSA and trifluoroacetate salt from the acetate salt can be attributed to a higher acidity of the acids.
After establishing the feasibility of the enone version of the intramolecular aza-[3 + 3] annulation we turned our attention to propyleine (12) (see Figure 2 and Scheme 3) as a possible target to showcase the new enone aza-[3 + 3] annulation. Propyleine (12) was isolated in 1972 from Propylaea quatuordecimpunctata in a continued effort by Tursch and co-workers [46,47] in their isolation of the azaphenalene family of defensive alkaloids from various ladybug beetles [38-45]. Mueller and Thompson [48] in 1980 found it interesting that the isomeric enamine named isopropyleine (14) was not reported in the original paper [46,47] taking into account that the isolation conditions involved acid–base extractions. It is conceivable that propyleine (12) and isopropyleine (14) could be interconvertable under acidic conditions via intermediate iminium salt 13.
![[1860-5397-9-131-i3]](/bjoc/content/inline/1860-5397-9-131-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Propyleine-isopropeleine interconversion.
Scheme 3: Propyleine-isopropeleine interconversion.
To investigate this matter, Mueller and Thompson carried out the first and the only synthesis of propyleine known to date [48]. They were able to take a mixture of propyleine and isopropyleine with 1:3 ratio as determined by 1H NMR and watch two sets of proton resonances collapse into one that corresponds to the iminium salt 13 after addition of TFA. This experiment strongly suggested that propyleine (12) and isopropyleine (14) could equilibrate under acidic conditions, thereby implying that the observed ratio of 12 and 14 represents a thermodynamic one in favor of the more stable isopropyleine [48]. With these experimental findings, Mueller and Thompson concluded that the alkaloid isolated by Tursch and co-workers [46,47] was in fact a mixture of readily interconvertable 12 and 14.
We found this controversy in the isolation and total synthesis papers an interesting one that deserves further investigations. Consequently, we performed ab initio calculations on 12 and 14 to gain insight into their relative thermodynamic stability. Models of the most stable conformers and their corresponding energies are shown in Figure 3. To our surprise, propyleine (12) is 2.59 kcal mol−1 more stable than isopropyleine (14), which is the opposite of the Mueller–Thompson postulation [48]. Our calculations suggest that the ratio obtained from the Mueller–Thompson study was likely determined by the kinetics (kB versus kR) in the deprotonation step or the tautormerization process from the iminium salt 13, and not by thermodynamics as originally proposed. Resolving this interesting literature controversy added extra incentive for us to pursue propyleine.
![[1860-5397-9-131-3]](/bjoc/content/figures/1860-5397-9-131-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Relative stabilities of propyleine and isopropyleine.
Figure 3: Relative stabilities of propyleine and isopropyleine.
Retrosynthetically, we envisioned propyleine (12) to come from the decarboxylation reaction of vinylogous carbamic acid 15 [10,28], which could be derived from stereoselective hydrogenation of the endocyclic olefin in tricycle 16 (Scheme 4). Vinylogous urethane 16 should be accessible via our intramolecular aza-[3 + 3] annulation of enone 17. Enone 17 could be E or Z with respect to the vinylogous urethane olefin, and cis or trans with respect to the enone double bond. Thus, from the spectroscopic analysis stand point, synthesis of 17 could be messy.
![[1860-5397-9-131-i4]](/bjoc/content/inline/1860-5397-9-131-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Retrosynthesis of propyleine (12).
Scheme 4: Retrosynthesis of propyleine (12).
In the forward direction to synthesize 17, we used the approach developed in our model study for the synthesis of precoccinelline, hippodamine and myrrhine alkaloids [30,31]. Specifically, TBDPS-protected 3-butyne-2-ol 18 was converted to bromoalkene 19 in two steps involving alkylation with an excess of dibromopropane and subsequent Lindlar’s hydrogenation (Scheme 5). Grignard addition to the glutarimide Mg-salt 20 followed by reduction of the Mg salt 21 afforded lactam 22 as an inseparable 1:1 mixture of two diastereomers in 77% overall yield [68-70].
![[1860-5397-9-131-i5]](/bjoc/content/inline/1860-5397-9-131-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Vinylogous amide 23 was prepared by O-methylation of lactam 22 with freshly distilled MeOTf followed by condensation with Meldrum’s acid in the presence of Ni(acac)2. Treatment of 23 with MeONa in MeOH under reflux gave vinylogous urethane 24. It is noteworthy that the geometry of the vinylogous urethane double bond exclusively favored Z, presumably due to the internal hydrogen bonding. Cleavage of the TBDPS group with TBAF provided allylic alcohol 25 in 79% from 23. However, submission of 25 to the standard MnO2 oxidation procedure did not lead to enone 17, with only the starting material being recovered (Table 2). After extensive screening of various oxidation protocols, we succeeded with the Doering–Parikh conditions [71], and the cis geometry of the enone olefin was preserved under these conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-131-i7.svg?max-width=637&scale=1.0)
With enone 17-cis in hand, we studied its intramolecular aza-[3 + 3] annulation reaction utilizing piperidinium salts (Table 3). When compound 17-cis was treated with the acetate salt in EtOAc at rt, no reaction was observed after 18 h (Table 3, entry 1). Heating this reaction mixture at 85 °C for 12 h only led to slow isomerization of cis-enone 17 to the thermodynamically more stable trans-isomer again with no formation of the desired annulation product 16 (Table 3, entry 2). The inefficiency of the piperidinium acetate salt in the intramolecular aza-[3 + 3] annulation of enones was consistent with our previous findings (see Table 1). We were also not successful in converting enone 17-cis to the desired tricycle 16 using the more reactive trifluoroacetate salt and EtOAc as the solvent. In this case also only isomerization to trans-enone was detected (Table 3, entry 3). Heating this reaction to 130 °C led to eventual decomposition of the starting material (Table 3, entry 4).
Table 3: Aza-[3 + 3] annulations of 17-cis.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-131-i8.svg?max-width=637&scale=1.0)
|
|||||
| entrya | HX (1.0 equiv) | solvent | temp (°C) | time (h) | yield (%)b |
|---|---|---|---|---|---|
| 1 | HOAc | EtOAc | 25 | 18 | – |
| 2 | HOAc | EtOAc | 85 | 12 | 17-trans |
| 3 | HO2CCF3 | EtOAc | 55 | 12 | 17-trans |
| 4 | HO2CCF3 | EtOAc | 130 | 12 | decomp |
| 5 | HO2CCF3 | toluene | 100 | 5 | 30% of 26 |
aAll reactions run in a sealed tube. Concn = 0.03–0.04 M; 5.0 equiv Na2SO4. bIsolated yields.
However, when enone 17-cis was heated in toluene at 100 °C, complete consumption of starting material was observed after 5 h (Table 3, entry 5). Even though the reaction was relatively messy, the formation of the desired cycloadduct 16 was confirmed by the presence of characteristic signals in the 1H NMR spectra of the crude mixture. Unfortunately, attempts to isolate the annulation product in pure form were not successful, probably due to the high instability of the electron-rich dihydropyridine moiety in 16. Based on our previous experience with precarious annulation products, in situ hydrogenation was then carried out, and we managed to track down ~30% of the desired reduced aza-annulation product 26, although stereoselectivity for the reduction was modest. We ultimately elected not to force our way toward propyleine using the enone aza-[3 + 3] annulation, as we succeeded in total syntheses of other members of the azaphenalene alkaloid family through annulations with enals [30,31]. While it is disappointing that this particular system may have lacked sufficient stability for this to be a suitable synthetic approach, success in an enone version of intramolecular aza-[3 + 3] annulation will allow us to find future applications.
Conclusion
Herein, we have described a successful enone version of intramolecular aza-[3 + 3] annulation reaction. Use of piperidinium trifluoroacetate salt as the catalyst and toluene as the solvent appears to be critical for a successful annulation. We also demonstrated for the first time that microwave irradiation can accelerate aza-[3 + 3] annulation reactions. An attempt to expand the scope of enone aza-[3 + 3] annulation was made in the form of propyleine synthesis as a proof of concept. While the synthesis of an enone annulation precursor was successfully accomplished, the annulation itself proved to be challenging and was only modestly successful. Future investigations are underway to pursue alkaloid synthesis via enone aza-[3 + 3] annulation.
Supporting Information
| Supporting Information File 1: Experimental section. | ||
| Format: PDF | Size: 698.7 KB | Download |
References
-
Harrity, J. P. A.; Provoost, O. Org. Biomol. Chem. 2005, 3, 1349. doi:10.1039/b502349c
See for a leading review on hetero-[3 +3] annulations.
Return to citation in text: [1] [2] [3] -
Hsung, R. P.; Kurdyumov, A. V.; Sydorenko, N. Eur. J. Org. Chem. 2005, 23. doi:10.1002/ejoc.200400567
See for a leading review on hetero-[3 +3] annulations.
Return to citation in text: [1] [2] -
Buchanan, G. S.; Feltenberger, J. B.; Hsung, R. P. Curr. Org. Synth. 2010, 7, 363. doi:10.2174/157017910791414490
See for a leading review on aza-[3 +3] annulations.
Return to citation in text: [1] [2] -
Lawrence, A. K.; Gademann, K. Synthesis 2008, 331. doi:10.1055/s-2008-1032134
See for a leading review on aza-[3 +3] annulations.
Return to citation in text: [1] [2] -
Sklenicka, H. M.; Shen, H. C.; Wei, L.-L.; Zehnder, L. R.; McLaughlin, M. J.; Hsung, R. P. Trends Heterocycl. Chem. 2001, 7, 1–24.
Return to citation in text: [1] -
Hsung, R. P.; Cole, K. P. The Total Synthesis of (-)-Arisugacin A. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Pergamon Press: Oxford, 2004; Vol. 4, pp 41–70.
Return to citation in text: [1] -
Tang, Y.; Oppenheimer, J.; Song, Z.; You, L.; Zhang, X.; Hsung, R. P. Tetrahedron 2006, 62, 10785. doi:10.1016/j.tet.2006.08.054
Return to citation in text: [1] -
Hsung, R. P.; Wei, L.-L.; Sklenicka, H. M.; Douglas, C. J.; McLaughlin, M. J.; Mulder, J. A.; Yao, L. J. Org. Lett. 1999, 1, 509. doi:10.1021/ol990107v
See for method development.
Return to citation in text: [1] [2] -
Sklenicka, H. M.; Hsung, R. P.; Wei, L.-L.; McLaughlin, M. J.; Gerasyuto, A. I.; Degen, S. J. Org. Lett. 2000, 2, 1161. doi:10.1021/ol000049+
Return to citation in text: [1] [2] -
McLaughlin, M. J.; Hsung, R. P.; Cole, K. C.; Hahn, J. M.; Wang, J. Org. Lett. 2002, 4, 2017. doi:10.1021/ol020052o
Return to citation in text: [1] [2] [3] -
Sydorenko, N.; Hsung, R. P.; Darwish, O. S.; Hahn, J. M.; Liu, J. J. Org. Chem. 2004, 69, 6732. doi:10.1021/jo049108d
Return to citation in text: [1] [2] -
Gerasyuto, A. I.; Hsung, R. P.; Sydorenko, N.; Slafer, B. W. J. Org. Chem. 2005, 70, 4248. doi:10.1021/jo050171s
Return to citation in text: [1] [2] [3] -
Sydorenko, N.; Hsung, R. P.; Vera, E. L. Org. Lett. 2006, 8, 2611. doi:10.1021/ol060932t
Return to citation in text: [1] [2] -
Ghosh, S. K.; Buchanan, G. S.; Long, Q. A.; Wei, Y.; Al-Rashid, Z. F.; Sklenicka, H. M.; Hsung, R. P. Tetrahedron 2008, 63, 883. doi:10.1016/j.tet.2007.09.089
Return to citation in text: [1] [2] -
Buchanan, G. S.; Dai, H.; Hsung, R. P.; Gerasyuto, A. I.; Schienebeck, C. M. Org. Lett. 2011, 13, 4402. doi:10.1021/ol2017438
Return to citation in text: [1] [2] -
Wei, L.-L.; Hsung, R. P.; Sklenicka, H. M.; Gerasyuto, A. I. Angew. Chem., Int. Ed. 2001, 40, 1516. doi:10.1002/1521-3773(20010417)40:8<1516::AID-ANIE1516>3.0.CO;2-V
See for investigations related to intramolecular annulations.
Return to citation in text: [1] [2] [3] [4] -
Sklenicka, H. M.; Hsung, R. P.; McLaughlin, M. J.; Wei, L.-L.; Gerasyuto, A. I.; Brennessel, W. W. J. Am. Chem. Soc. 2002, 124, 10435. doi:10.1021/ja020698b
Return to citation in text: [1] [2] -
Iaroshenko, V. O.; Ali, S.; Babar, T. M.; Dudkin, S.; Mkrtchyan, S.; Rama, N. H.; Villinge, A.; Langer, P. Tetrahedron Lett. 2011, 52, 373. doi:10.1016/j.tetlet.2010.11.028
See for recent studies in aza-annulations.
Return to citation in text: [1] -
Guo, H.; Xu, Q.; Kwon, O. J. Am. Chem. Soc. 2009, 131, 6318. doi:10.1021/ja8097349
Return to citation in text: [1] -
Jeanne Alladoum, J.; Toum, V.; Hebbe, S.; Kadouri-Puchot, C.; Dechoux, L. Tetrahedron Lett. 2009, 50, 617. doi:10.1016/j.tetlet.2008.11.070
Return to citation in text: [1] -
Zhong, W.; Lin, F.; Chen, R.; Su, W. Synthesis 2008, 2561. doi:10.1055/s-2008-1078601
Return to citation in text: [1] -
Hayashi, Y.; Gotoh, H.; Masui, R.; Ishikawa, H. Angew. Chem., Int. Ed. 2008, 47, 4012. doi:10.1002/anie.200800662
Return to citation in text: [1] -
Mancey, N. C.; Butlin, R. J.; Harrity, J. P. A. Synlett 2008, 2647. doi:10.1055/s-0028-1083525
Return to citation in text: [1] -
Trost, B. M.; Dong, G. Org. Lett. 2007, 9, 2357. doi:10.1021/ol070742y
Return to citation in text: [1] -
Provoost, O. Y.; Hazelwood, A. J.; Harrity, J. P. A. Beilstein J. Org. Chem. 2007, 3, No. 8. doi:10.1186/1860-5397-3-8
Return to citation in text: [1] -
Luo, S.; Zificsak, C. A.; Hsung, R. P. Org. Lett. 2003, 5, 4709. doi:10.1021/ol030114q
See for natural product synthesis using the intramolecular annulation.
Return to citation in text: [1] [2] [3] -
Sydorenko, N.; Zificsak, C. A.; Gerasyuto, A. I.; Hsung, R. P. Org. Biomol. Chem. 2005, 3, 2140. doi:10.1039/b503862f
Return to citation in text: [1] [2] [3] -
Swidorski, J. J.; Wang, J.; Hsung, R. P. Org. Lett. 2006, 8, 777. doi:10.1021/ol053059p
Return to citation in text: [1] [2] [3] [4] -
Wang, J.; Swidorski, J. J.; Sydorenko, N.; Hsung, R. P.; Coverdale, H. A.; Kuyava, J. M.; Liu, J. Heterocycles 2006, 70, 423. doi:10.3987/COM-06-S(W)40
Return to citation in text: [1] [2] [3] -
Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Zhang, Y.; Gerasyuto, A. I.; Long, Q. A.; Hsung, R. P. Synlett 2009, 237. doi:10.1055/s-0028-1087661
Return to citation in text: [1] [2] [3] -
Robinson, R. J. Chem. Soc. 1917, 762.
Return to citation in text: [1] -
Boger, D. L.; Patel, M. Progress in Heterocyclic Chemistry; Suschitzky. H.; Scriven, E. F. V., Eds.; Pergamon: London, 1989, Vol. 1, pp. 30 ff. See for a leading review.
Return to citation in text: [1] -
Boger, D. L. Chem. Rev. 1986, 86, 781. doi:10.1021/cr00075a004
Return to citation in text: [1] -
Boger, D. L. Tetrahedron 1983, 39, 2869. doi:10.1016/S0040-4020(01)92154-4
Return to citation in text: [1] -
Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press, 1991; Vol. 4 and 5.
See chapters on cycloaddition reactions.
Return to citation in text: [1] -
Happ, G. M.; Eisner, T. Science 1961, 134, 329. doi:10.1126/science.134.3475.329
See for leading reviews on azaphenalene alkaloids.
Return to citation in text: [1] [2] -
King, A. G.; Meinwald, J. Chem. Rev. 1996, 96, 1105. doi:10.1021/cr950242v
Return to citation in text: [1] [2] -
Stevens, R. V. Acc. Chem. Res. 1984, 17, 289. doi:10.1021/ar00104a005
Return to citation in text: [1] [2] -
Tursch, B.; Daloze, D.; Dupont, M.; Pasteels, J. M.; Tricot, M.-C. Experientia 1971, 27, 1380. doi:10.1007/BF02154239
See for isolations of azaphenalene alkaloids.
Return to citation in text: [1] [2] -
Karlsson, R.; Losman, D. J. Chem. Soc., Chem. Commun. 1972, 626. doi:10.1039/C39720000626
Return to citation in text: [1] [2] -
Tursch, B.; Daloze, D.; Pasteels, J. M.; Cravador, A.; Braekman, J. C.; Hootele, C.; Zimmermann, D. Bull. Soc. Chim. Belg. 1972, 81, 649.
Return to citation in text: [1] [2] -
Tursch, B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Cravador, A.; Losman, D.; Karlsson, R. Tetrahedron Lett. 1974, 409. doi:10.1016/S0040-4039(01)82228-0
Return to citation in text: [1] [2] -
Tursch, B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Pasteels, J. M. Tetrahedron 1975, 31, 1541. doi:10.1016/0040-4020(75)87008-6
Return to citation in text: [1] [2] -
Tursch, B.; Daloze, D.; Hootele, C. Chimia 1972, 26, 74.
See for isolation of propyleine.
Return to citation in text: [1] [2] [3] [4] -
Moore, B. P.; Brown, W. V. Insect Biochem. 1978, 8, 393. doi:10.1016/0020-1790(78)90027-6
Return to citation in text: [1] [2] [3] [4] -
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ma, X.; Gang, D. R. Nat. Prod. Rep. 2004, 21, 752. doi:10.1039/b409720n
See for leading reviews on lycopodium alkaloids.
Return to citation in text: [1] -
Ayer, W. A. Nat. Prod. Rep. 1991, 8, 455. doi:10.1039/np9910800455
Return to citation in text: [1] -
MacLean, D. B. In The Alkaloids; Brossi, A., Ed.; Academic Press: New York, 1985; Vol. 26, p 241.
Return to citation in text: [1] -
Henry, T. A. The Plant Alkaloids, 4th ed.; The Blackiston Company, 1949; pp 394 ff.
Return to citation in text: [1] [2] [3] [4] -
Shamma, M. In The Isoquinoline Alkaloids. Chemistry and Pharmacology; Blomquist, A. T.; Wasserman, H., Eds.; Organic Chemistry, Vol. 25; Academic Press: New York, 1972; pp 427–455.
Return to citation in text: [1] [2] [3] [4] -
Brown, R. T. In The Chemistry and Biology of Isoquinoline Alkaloids; Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds.; Springer-Verlag: Berlin, 1985; pp 205–207.
Return to citation in text: [1] [2] [3] [4] -
Szántay, C. In The Alkaloids. Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: London, 1998; Vol. 50, pp 379–380.
Return to citation in text: [1] [2] [3] [4] -
Rapoport, H.; Windgassen, R. J.; Hughes, N. A.; Onak, T. P. J. Am. Chem. Soc. 1960, 82, 4404. doi:10.1021/ja01501a069
Return to citation in text: [1] -
Janot, M.-M. Tetrahedron 1961, 14, 113. doi:10.1016/0040-4020(61)80089-6
Return to citation in text: [1] -
Wiegrebe, W.; Kramer, W. J.; Shamma, M. J. Nat. Prod. 1984, 47, 397. doi:10.1021/np50033a001
Return to citation in text: [1] [2] -
Piso, W.; Marcgraf, G. Historia Naturalis Brasiliae, Elzevir, L. Ed.; Amsterdam, 1648; Ch. 65, De Ipecacuanha, eiusque facultatibus, pp 101–102.
Return to citation in text: [1] [2] -
Wild, R. B. The Pharmacology of ipecacuanha Alkaloids. In The Western Druggist; G. P. Engelhardt and Co.: Chicago, 1897; Vol. 18, p 12.
Return to citation in text: [1] [2] -
Lloyd, U. J. Cephaelis ipecacuanha. In The Western Druggist, Chicago, August, 1897; Vol. 19, pp 346–350.
Return to citation in text: [1] [2] -
The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 12th ed.; Budavari, S.; O’Neil, M. J.; Smith, A.; Heckelman, P. E.; Kinneary, J. F., Eds.; Merck and Co. Inc.: Whitehouse Station, NJ, 1996; pp 3600–3601.
Return to citation in text: [1] -
Zimmerman, J. L.; Rudis, M. Chapter 73. In Critical Care Medicine. Principles of Diagnosis and Management in the Adult, 2nd ed.; Parrillo, J. E., Ed.; Mosby, Inc.: St. Louis, MO, 2002; pp 1506–1507.
Return to citation in text: [1] -
Ma, W.-W.; Anderson, J. E.; McKenzie, A. T.; Byrn, S. R.; McLaughlin, J. L.; Hudson, M. S. J. Nat. Prod. 1990, 53, 1009. doi:10.1021/np50070a041
Return to citation in text: [1] -
A mixture of EtOAc and toluene was employed to allow for both complete dissolution of the substrate and heating at higher temperatures.
Return to citation in text: [1] -
Bose, D. S.; Kumar, R. K. Heterocycles 2006, 68, 549. doi:10.3987/COM-06-10668
See for an account on the use of microwaves in aza-[3 + 3] annulations.
Return to citation in text: [1] -
Kang, Y.; Mei, Y.; Du, Y.; Jin, Z. Org. Lett. 2003, 5, 4481. doi:10.1021/ol030109m
See for an account on the use of microwaves in oxa-[3 + 3] annulations.
Return to citation in text: [1] -
Karstens, W. F. J.; Stol, M.; Rutjes, F. P. J. T.; Kooijman, H.; Spek, A. L.; Hiemstra, H. J. Organomet. Chem. 2001, 624, 244. doi:10.1016/S0022-328X(00)00935-9
Return to citation in text: [1] -
Esch, P. M.; Hiemstra, H.; Klaver, W. J.; Speckamp, W. N. Heterocycles 1987, 26, 75. doi:10.3987/R-1987-01-0075
Return to citation in text: [1] -
Evans, D. A.; Thomas, E. W.; Cherpeck, R. E. J. Am. Chem. Soc. 1982, 104, 3695. doi:10.1021/ja00377a026
Return to citation in text: [1] -
Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505. doi:10.1021/ja00997a067
Return to citation in text: [1]
| 10. | McLaughlin, M. J.; Hsung, R. P.; Cole, K. C.; Hahn, J. M.; Wang, J. Org. Lett. 2002, 4, 2017. doi:10.1021/ol020052o |
| 28. | Swidorski, J. J.; Wang, J.; Hsung, R. P. Org. Lett. 2006, 8, 777. doi:10.1021/ol053059p |
| 30. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359 |
| 31. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h |
| 68. | Karstens, W. F. J.; Stol, M.; Rutjes, F. P. J. T.; Kooijman, H.; Spek, A. L.; Hiemstra, H. J. Organomet. Chem. 2001, 624, 244. doi:10.1016/S0022-328X(00)00935-9 |
| 69. | Esch, P. M.; Hiemstra, H.; Klaver, W. J.; Speckamp, W. N. Heterocycles 1987, 26, 75. doi:10.3987/R-1987-01-0075 |
| 70. | Evans, D. A.; Thomas, E. W.; Cherpeck, R. E. J. Am. Chem. Soc. 1982, 104, 3695. doi:10.1021/ja00377a026 |
| 1. |
Harrity, J. P. A.; Provoost, O. Org. Biomol. Chem. 2005, 3, 1349. doi:10.1039/b502349c
See for a leading review on hetero-[3 +3] annulations. |
| 2. |
Hsung, R. P.; Kurdyumov, A. V.; Sydorenko, N. Eur. J. Org. Chem. 2005, 23. doi:10.1002/ejoc.200400567
See for a leading review on hetero-[3 +3] annulations. |
| 3. |
Buchanan, G. S.; Feltenberger, J. B.; Hsung, R. P. Curr. Org. Synth. 2010, 7, 363. doi:10.2174/157017910791414490
See for a leading review on aza-[3 +3] annulations. |
| 4. |
Lawrence, A. K.; Gademann, K. Synthesis 2008, 331. doi:10.1055/s-2008-1032134
See for a leading review on aza-[3 +3] annulations. |
| 5. | Sklenicka, H. M.; Shen, H. C.; Wei, L.-L.; Zehnder, L. R.; McLaughlin, M. J.; Hsung, R. P. Trends Heterocycl. Chem. 2001, 7, 1–24. |
| 6. | Hsung, R. P.; Cole, K. P. The Total Synthesis of (-)-Arisugacin A. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Pergamon Press: Oxford, 2004; Vol. 4, pp 41–70. |
| 7. | Tang, Y.; Oppenheimer, J.; Song, Z.; You, L.; Zhang, X.; Hsung, R. P. Tetrahedron 2006, 62, 10785. doi:10.1016/j.tet.2006.08.054 |
| 8. |
Hsung, R. P.; Wei, L.-L.; Sklenicka, H. M.; Douglas, C. J.; McLaughlin, M. J.; Mulder, J. A.; Yao, L. J. Org. Lett. 1999, 1, 509. doi:10.1021/ol990107v
See for method development. |
| 9. | Sklenicka, H. M.; Hsung, R. P.; Wei, L.-L.; McLaughlin, M. J.; Gerasyuto, A. I.; Degen, S. J. Org. Lett. 2000, 2, 1161. doi:10.1021/ol000049+ |
| 10. | McLaughlin, M. J.; Hsung, R. P.; Cole, K. C.; Hahn, J. M.; Wang, J. Org. Lett. 2002, 4, 2017. doi:10.1021/ol020052o |
| 11. | Sydorenko, N.; Hsung, R. P.; Darwish, O. S.; Hahn, J. M.; Liu, J. J. Org. Chem. 2004, 69, 6732. doi:10.1021/jo049108d |
| 12. | Gerasyuto, A. I.; Hsung, R. P.; Sydorenko, N.; Slafer, B. W. J. Org. Chem. 2005, 70, 4248. doi:10.1021/jo050171s |
| 13. | Sydorenko, N.; Hsung, R. P.; Vera, E. L. Org. Lett. 2006, 8, 2611. doi:10.1021/ol060932t |
| 14. | Ghosh, S. K.; Buchanan, G. S.; Long, Q. A.; Wei, Y.; Al-Rashid, Z. F.; Sklenicka, H. M.; Hsung, R. P. Tetrahedron 2008, 63, 883. doi:10.1016/j.tet.2007.09.089 |
| 15. | Buchanan, G. S.; Dai, H.; Hsung, R. P.; Gerasyuto, A. I.; Schienebeck, C. M. Org. Lett. 2011, 13, 4402. doi:10.1021/ol2017438 |
| 16. |
Wei, L.-L.; Hsung, R. P.; Sklenicka, H. M.; Gerasyuto, A. I. Angew. Chem., Int. Ed. 2001, 40, 1516. doi:10.1002/1521-3773(20010417)40:8<1516::AID-ANIE1516>3.0.CO;2-V
See for investigations related to intramolecular annulations. |
| 17. | Sklenicka, H. M.; Hsung, R. P.; McLaughlin, M. J.; Wei, L.-L.; Gerasyuto, A. I.; Brennessel, W. W. J. Am. Chem. Soc. 2002, 124, 10435. doi:10.1021/ja020698b |
| 18. |
Iaroshenko, V. O.; Ali, S.; Babar, T. M.; Dudkin, S.; Mkrtchyan, S.; Rama, N. H.; Villinge, A.; Langer, P. Tetrahedron Lett. 2011, 52, 373. doi:10.1016/j.tetlet.2010.11.028
See for recent studies in aza-annulations. |
| 19. | Guo, H.; Xu, Q.; Kwon, O. J. Am. Chem. Soc. 2009, 131, 6318. doi:10.1021/ja8097349 |
| 20. | Jeanne Alladoum, J.; Toum, V.; Hebbe, S.; Kadouri-Puchot, C.; Dechoux, L. Tetrahedron Lett. 2009, 50, 617. doi:10.1016/j.tetlet.2008.11.070 |
| 21. | Zhong, W.; Lin, F.; Chen, R.; Su, W. Synthesis 2008, 2561. doi:10.1055/s-2008-1078601 |
| 22. | Hayashi, Y.; Gotoh, H.; Masui, R.; Ishikawa, H. Angew. Chem., Int. Ed. 2008, 47, 4012. doi:10.1002/anie.200800662 |
| 23. | Mancey, N. C.; Butlin, R. J.; Harrity, J. P. A. Synlett 2008, 2647. doi:10.1055/s-0028-1083525 |
| 24. | Trost, B. M.; Dong, G. Org. Lett. 2007, 9, 2357. doi:10.1021/ol070742y |
| 25. | Provoost, O. Y.; Hazelwood, A. J.; Harrity, J. P. A. Beilstein J. Org. Chem. 2007, 3, No. 8. doi:10.1186/1860-5397-3-8 |
| 26. |
Luo, S.; Zificsak, C. A.; Hsung, R. P. Org. Lett. 2003, 5, 4709. doi:10.1021/ol030114q
See for natural product synthesis using the intramolecular annulation. |
| 27. | Sydorenko, N.; Zificsak, C. A.; Gerasyuto, A. I.; Hsung, R. P. Org. Biomol. Chem. 2005, 3, 2140. doi:10.1039/b503862f |
| 28. | Swidorski, J. J.; Wang, J.; Hsung, R. P. Org. Lett. 2006, 8, 777. doi:10.1021/ol053059p |
| 29. | Wang, J.; Swidorski, J. J.; Sydorenko, N.; Hsung, R. P.; Coverdale, H. A.; Kuyava, J. M.; Liu, J. Heterocycles 2006, 70, 423. doi:10.3987/COM-06-S(W)40 |
| 30. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359 |
| 31. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h |
| 32. | Zhang, Y.; Gerasyuto, A. I.; Long, Q. A.; Hsung, R. P. Synlett 2009, 237. doi:10.1055/s-0028-1087661 |
| 1. |
Harrity, J. P. A.; Provoost, O. Org. Biomol. Chem. 2005, 3, 1349. doi:10.1039/b502349c
See for a leading review on hetero-[3 +3] annulations. |
| 2. |
Hsung, R. P.; Kurdyumov, A. V.; Sydorenko, N. Eur. J. Org. Chem. 2005, 23. doi:10.1002/ejoc.200400567
See for a leading review on hetero-[3 +3] annulations. |
| 3. |
Buchanan, G. S.; Feltenberger, J. B.; Hsung, R. P. Curr. Org. Synth. 2010, 7, 363. doi:10.2174/157017910791414490
See for a leading review on aza-[3 +3] annulations. |
| 4. |
Lawrence, A. K.; Gademann, K. Synthesis 2008, 331. doi:10.1055/s-2008-1032134
See for a leading review on aza-[3 +3] annulations. |
| 8. |
Hsung, R. P.; Wei, L.-L.; Sklenicka, H. M.; Douglas, C. J.; McLaughlin, M. J.; Mulder, J. A.; Yao, L. J. Org. Lett. 1999, 1, 509. doi:10.1021/ol990107v
See for method development. |
| 9. | Sklenicka, H. M.; Hsung, R. P.; Wei, L.-L.; McLaughlin, M. J.; Gerasyuto, A. I.; Degen, S. J. Org. Lett. 2000, 2, 1161. doi:10.1021/ol000049+ |
| 10. | McLaughlin, M. J.; Hsung, R. P.; Cole, K. C.; Hahn, J. M.; Wang, J. Org. Lett. 2002, 4, 2017. doi:10.1021/ol020052o |
| 11. | Sydorenko, N.; Hsung, R. P.; Darwish, O. S.; Hahn, J. M.; Liu, J. J. Org. Chem. 2004, 69, 6732. doi:10.1021/jo049108d |
| 12. | Gerasyuto, A. I.; Hsung, R. P.; Sydorenko, N.; Slafer, B. W. J. Org. Chem. 2005, 70, 4248. doi:10.1021/jo050171s |
| 13. | Sydorenko, N.; Hsung, R. P.; Vera, E. L. Org. Lett. 2006, 8, 2611. doi:10.1021/ol060932t |
| 14. | Ghosh, S. K.; Buchanan, G. S.; Long, Q. A.; Wei, Y.; Al-Rashid, Z. F.; Sklenicka, H. M.; Hsung, R. P. Tetrahedron 2008, 63, 883. doi:10.1016/j.tet.2007.09.089 |
| 15. | Buchanan, G. S.; Dai, H.; Hsung, R. P.; Gerasyuto, A. I.; Schienebeck, C. M. Org. Lett. 2011, 13, 4402. doi:10.1021/ol2017438 |
| 52. | Henry, T. A. The Plant Alkaloids, 4th ed.; The Blackiston Company, 1949; pp 394 ff. |
| 53. | Shamma, M. In The Isoquinoline Alkaloids. Chemistry and Pharmacology; Blomquist, A. T.; Wasserman, H., Eds.; Organic Chemistry, Vol. 25; Academic Press: New York, 1972; pp 427–455. |
| 54. | Brown, R. T. In The Chemistry and Biology of Isoquinoline Alkaloids; Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds.; Springer-Verlag: Berlin, 1985; pp 205–207. |
| 55. | Szántay, C. In The Alkaloids. Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: London, 1998; Vol. 50, pp 379–380. |
| 58. | Wiegrebe, W.; Kramer, W. J.; Shamma, M. J. Nat. Prod. 1984, 47, 397. doi:10.1021/np50033a001 |
| 59. | Piso, W.; Marcgraf, G. Historia Naturalis Brasiliae, Elzevir, L. Ed.; Amsterdam, 1648; Ch. 65, De Ipecacuanha, eiusque facultatibus, pp 101–102. |
| 60. | Wild, R. B. The Pharmacology of ipecacuanha Alkaloids. In The Western Druggist; G. P. Engelhardt and Co.: Chicago, 1897; Vol. 18, p 12. |
| 61. | Lloyd, U. J. Cephaelis ipecacuanha. In The Western Druggist, Chicago, August, 1897; Vol. 19, pp 346–350. |
| 62. | The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 12th ed.; Budavari, S.; O’Neil, M. J.; Smith, A.; Heckelman, P. E.; Kinneary, J. F., Eds.; Merck and Co. Inc.: Whitehouse Station, NJ, 1996; pp 3600–3601. |
| 63. | Zimmerman, J. L.; Rudis, M. Chapter 73. In Critical Care Medicine. Principles of Diagnosis and Management in the Adult, 2nd ed.; Parrillo, J. E., Ed.; Mosby, Inc.: St. Louis, MO, 2002; pp 1506–1507. |
| 34. | Boger, D. L.; Patel, M. Progress in Heterocyclic Chemistry; Suschitzky. H.; Scriven, E. F. V., Eds.; Pergamon: London, 1989, Vol. 1, pp. 30 ff. See for a leading review. |
| 35. | Boger, D. L. Chem. Rev. 1986, 86, 781. doi:10.1021/cr00075a004 |
| 36. | Boger, D. L. Tetrahedron 1983, 39, 2869. doi:10.1016/S0040-4020(01)92154-4 |
| 37. |
Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press, 1991; Vol. 4 and 5.
See chapters on cycloaddition reactions. |
| 64. | Ma, W.-W.; Anderson, J. E.; McKenzie, A. T.; Byrn, S. R.; McLaughlin, J. L.; Hudson, M. S. J. Nat. Prod. 1990, 53, 1009. doi:10.1021/np50070a041 |
| 52. | Henry, T. A. The Plant Alkaloids, 4th ed.; The Blackiston Company, 1949; pp 394 ff. |
| 53. | Shamma, M. In The Isoquinoline Alkaloids. Chemistry and Pharmacology; Blomquist, A. T.; Wasserman, H., Eds.; Organic Chemistry, Vol. 25; Academic Press: New York, 1972; pp 427–455. |
| 54. | Brown, R. T. In The Chemistry and Biology of Isoquinoline Alkaloids; Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds.; Springer-Verlag: Berlin, 1985; pp 205–207. |
| 55. | Szántay, C. In The Alkaloids. Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: London, 1998; Vol. 50, pp 379–380. |
| 56. | Rapoport, H.; Windgassen, R. J.; Hughes, N. A.; Onak, T. P. J. Am. Chem. Soc. 1960, 82, 4404. doi:10.1021/ja01501a069 |
| 57. | Janot, M.-M. Tetrahedron 1961, 14, 113. doi:10.1016/0040-4020(61)80089-6 |
| 1. |
Harrity, J. P. A.; Provoost, O. Org. Biomol. Chem. 2005, 3, 1349. doi:10.1039/b502349c
See for a leading review on hetero-[3 +3] annulations. |
| 48. |
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine. |
| 52. | Henry, T. A. The Plant Alkaloids, 4th ed.; The Blackiston Company, 1949; pp 394 ff. |
| 53. | Shamma, M. In The Isoquinoline Alkaloids. Chemistry and Pharmacology; Blomquist, A. T.; Wasserman, H., Eds.; Organic Chemistry, Vol. 25; Academic Press: New York, 1972; pp 427–455. |
| 54. | Brown, R. T. In The Chemistry and Biology of Isoquinoline Alkaloids; Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds.; Springer-Verlag: Berlin, 1985; pp 205–207. |
| 55. | Szántay, C. In The Alkaloids. Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: London, 1998; Vol. 50, pp 379–380. |
| 58. | Wiegrebe, W.; Kramer, W. J.; Shamma, M. J. Nat. Prod. 1984, 47, 397. doi:10.1021/np50033a001 |
| 59. | Piso, W.; Marcgraf, G. Historia Naturalis Brasiliae, Elzevir, L. Ed.; Amsterdam, 1648; Ch. 65, De Ipecacuanha, eiusque facultatibus, pp 101–102. |
| 60. | Wild, R. B. The Pharmacology of ipecacuanha Alkaloids. In The Western Druggist; G. P. Engelhardt and Co.: Chicago, 1897; Vol. 18, p 12. |
| 61. | Lloyd, U. J. Cephaelis ipecacuanha. In The Western Druggist, Chicago, August, 1897; Vol. 19, pp 346–350. |
| 30. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359 |
| 31. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h |
| 38. |
Happ, G. M.; Eisner, T. Science 1961, 134, 329. doi:10.1126/science.134.3475.329
See for leading reviews on azaphenalene alkaloids. |
| 39. | King, A. G.; Meinwald, J. Chem. Rev. 1996, 96, 1105. doi:10.1021/cr950242v |
| 40. | Stevens, R. V. Acc. Chem. Res. 1984, 17, 289. doi:10.1021/ar00104a005 |
| 41. |
Tursch, B.; Daloze, D.; Dupont, M.; Pasteels, J. M.; Tricot, M.-C. Experientia 1971, 27, 1380. doi:10.1007/BF02154239
See for isolations of azaphenalene alkaloids. |
| 42. | Karlsson, R.; Losman, D. J. Chem. Soc., Chem. Commun. 1972, 626. doi:10.1039/C39720000626 |
| 43. | Tursch, B.; Daloze, D.; Pasteels, J. M.; Cravador, A.; Braekman, J. C.; Hootele, C.; Zimmermann, D. Bull. Soc. Chim. Belg. 1972, 81, 649. |
| 44. | Tursch, B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Cravador, A.; Losman, D.; Karlsson, R. Tetrahedron Lett. 1974, 409. doi:10.1016/S0040-4039(01)82228-0 |
| 45. | Tursch, B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Pasteels, J. M. Tetrahedron 1975, 31, 1541. doi:10.1016/0040-4020(75)87008-6 |
| 49. |
Ma, X.; Gang, D. R. Nat. Prod. Rep. 2004, 21, 752. doi:10.1039/b409720n
See for leading reviews on lycopodium alkaloids. |
| 50. | Ayer, W. A. Nat. Prod. Rep. 1991, 8, 455. doi:10.1039/np9910800455 |
| 51. | MacLean, D. B. In The Alkaloids; Brossi, A., Ed.; Academic Press: New York, 1985; Vol. 26, p 241. |
| 16. |
Wei, L.-L.; Hsung, R. P.; Sklenicka, H. M.; Gerasyuto, A. I. Angew. Chem., Int. Ed. 2001, 40, 1516. doi:10.1002/1521-3773(20010417)40:8<1516::AID-ANIE1516>3.0.CO;2-V
See for investigations related to intramolecular annulations. |
| 26. |
Luo, S.; Zificsak, C. A.; Hsung, R. P. Org. Lett. 2003, 5, 4709. doi:10.1021/ol030114q
See for natural product synthesis using the intramolecular annulation. |
| 27. | Sydorenko, N.; Zificsak, C. A.; Gerasyuto, A. I.; Hsung, R. P. Org. Biomol. Chem. 2005, 3, 2140. doi:10.1039/b503862f |
| 28. | Swidorski, J. J.; Wang, J.; Hsung, R. P. Org. Lett. 2006, 8, 777. doi:10.1021/ol053059p |
| 29. | Wang, J.; Swidorski, J. J.; Sydorenko, N.; Hsung, R. P.; Coverdale, H. A.; Kuyava, J. M.; Liu, J. Heterocycles 2006, 70, 423. doi:10.3987/COM-06-S(W)40 |
| 30. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359 |
| 31. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h |
| 32. | Zhang, Y.; Gerasyuto, A. I.; Long, Q. A.; Hsung, R. P. Synlett 2009, 237. doi:10.1055/s-0028-1087661 |
| 52. | Henry, T. A. The Plant Alkaloids, 4th ed.; The Blackiston Company, 1949; pp 394 ff. |
| 53. | Shamma, M. In The Isoquinoline Alkaloids. Chemistry and Pharmacology; Blomquist, A. T.; Wasserman, H., Eds.; Organic Chemistry, Vol. 25; Academic Press: New York, 1972; pp 427–455. |
| 54. | Brown, R. T. In The Chemistry and Biology of Isoquinoline Alkaloids; Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds.; Springer-Verlag: Berlin, 1985; pp 205–207. |
| 55. | Szántay, C. In The Alkaloids. Chemistry and Biology; Cordell, G. A., Ed.; Academic Press: London, 1998; Vol. 50, pp 379–380. |
| 16. |
Wei, L.-L.; Hsung, R. P.; Sklenicka, H. M.; Gerasyuto, A. I. Angew. Chem., Int. Ed. 2001, 40, 1516. doi:10.1002/1521-3773(20010417)40:8<1516::AID-ANIE1516>3.0.CO;2-V
See for investigations related to intramolecular annulations. |
| 17. | Sklenicka, H. M.; Hsung, R. P.; McLaughlin, M. J.; Wei, L.-L.; Gerasyuto, A. I.; Brennessel, W. W. J. Am. Chem. Soc. 2002, 124, 10435. doi:10.1021/ja020698b |
| 71. | Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505. doi:10.1021/ja00997a067 |
| 16. |
Wei, L.-L.; Hsung, R. P.; Sklenicka, H. M.; Gerasyuto, A. I. Angew. Chem., Int. Ed. 2001, 40, 1516. doi:10.1002/1521-3773(20010417)40:8<1516::AID-ANIE1516>3.0.CO;2-V
See for investigations related to intramolecular annulations. |
| 26. |
Luo, S.; Zificsak, C. A.; Hsung, R. P. Org. Lett. 2003, 5, 4709. doi:10.1021/ol030114q
See for natural product synthesis using the intramolecular annulation. |
| 27. | Sydorenko, N.; Zificsak, C. A.; Gerasyuto, A. I.; Hsung, R. P. Org. Biomol. Chem. 2005, 3, 2140. doi:10.1039/b503862f |
| 28. | Swidorski, J. J.; Wang, J.; Hsung, R. P. Org. Lett. 2006, 8, 777. doi:10.1021/ol053059p |
| 29. | Wang, J.; Swidorski, J. J.; Sydorenko, N.; Hsung, R. P.; Coverdale, H. A.; Kuyava, J. M.; Liu, J. Heterocycles 2006, 70, 423. doi:10.3987/COM-06-S(W)40 |
| 30. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359 |
| 31. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h |
| 32. | Zhang, Y.; Gerasyuto, A. I.; Long, Q. A.; Hsung, R. P. Synlett 2009, 237. doi:10.1055/s-0028-1087661 |
| 46. |
Tursch, B.; Daloze, D.; Hootele, C. Chimia 1972, 26, 74.
See for isolation of propyleine. |
| 47. | Moore, B. P.; Brown, W. V. Insect Biochem. 1978, 8, 393. doi:10.1016/0020-1790(78)90027-6 |
| 48. |
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine. |
| 30. | Gerasyuto, A. I.; Hsung, R. P. Org. Lett. 2006, 8, 4899. doi:10.1021/ol0619359 |
| 31. | Gerasyuto, A. I.; Hsung, R. P. J. Org. Chem. 2007, 72, 2476. doi:10.1021/jo062533h |
| 12. | Gerasyuto, A. I.; Hsung, R. P.; Sydorenko, N.; Slafer, B. W. J. Org. Chem. 2005, 70, 4248. doi:10.1021/jo050171s |
| 65. | A mixture of EtOAc and toluene was employed to allow for both complete dissolution of the substrate and heating at higher temperatures. |
| 66. |
Bose, D. S.; Kumar, R. K. Heterocycles 2006, 68, 549. doi:10.3987/COM-06-10668
See for an account on the use of microwaves in aza-[3 + 3] annulations. |
| 67. |
Kang, Y.; Mei, Y.; Du, Y.; Jin, Z. Org. Lett. 2003, 5, 4481. doi:10.1021/ol030109m
See for an account on the use of microwaves in oxa-[3 + 3] annulations. |
| 46. |
Tursch, B.; Daloze, D.; Hootele, C. Chimia 1972, 26, 74.
See for isolation of propyleine. |
| 47. | Moore, B. P.; Brown, W. V. Insect Biochem. 1978, 8, 393. doi:10.1016/0020-1790(78)90027-6 |
| 48. |
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine. |
| 48. |
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine. |
| 48. |
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine. |
| 48. |
Mueller, R. H.; Thompson, M. E. Tetrahedron Lett. 1980, 21, 1097. doi:10.1016/S0040-4039(01)83923-X
See for a total synthesis of propyleine. |
| 46. |
Tursch, B.; Daloze, D.; Hootele, C. Chimia 1972, 26, 74.
See for isolation of propyleine. |
| 47. | Moore, B. P.; Brown, W. V. Insect Biochem. 1978, 8, 393. doi:10.1016/0020-1790(78)90027-6 |
| 46. |
Tursch, B.; Daloze, D.; Hootele, C. Chimia 1972, 26, 74.
See for isolation of propyleine. |
| 47. | Moore, B. P.; Brown, W. V. Insect Biochem. 1978, 8, 393. doi:10.1016/0020-1790(78)90027-6 |
| 38. |
Happ, G. M.; Eisner, T. Science 1961, 134, 329. doi:10.1126/science.134.3475.329
See for leading reviews on azaphenalene alkaloids. |
| 39. | King, A. G.; Meinwald, J. Chem. Rev. 1996, 96, 1105. doi:10.1021/cr950242v |
| 40. | Stevens, R. V. Acc. Chem. Res. 1984, 17, 289. doi:10.1021/ar00104a005 |
| 41. |
Tursch, B.; Daloze, D.; Dupont, M.; Pasteels, J. M.; Tricot, M.-C. Experientia 1971, 27, 1380. doi:10.1007/BF02154239
See for isolations of azaphenalene alkaloids. |
| 42. | Karlsson, R.; Losman, D. J. Chem. Soc., Chem. Commun. 1972, 626. doi:10.1039/C39720000626 |
| 43. | Tursch, B.; Daloze, D.; Pasteels, J. M.; Cravador, A.; Braekman, J. C.; Hootele, C.; Zimmermann, D. Bull. Soc. Chim. Belg. 1972, 81, 649. |
| 44. | Tursch, B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Cravador, A.; Losman, D.; Karlsson, R. Tetrahedron Lett. 1974, 409. doi:10.1016/S0040-4039(01)82228-0 |
| 45. | Tursch, B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Pasteels, J. M. Tetrahedron 1975, 31, 1541. doi:10.1016/0040-4020(75)87008-6 |
© 2013 Gerasyuto et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)