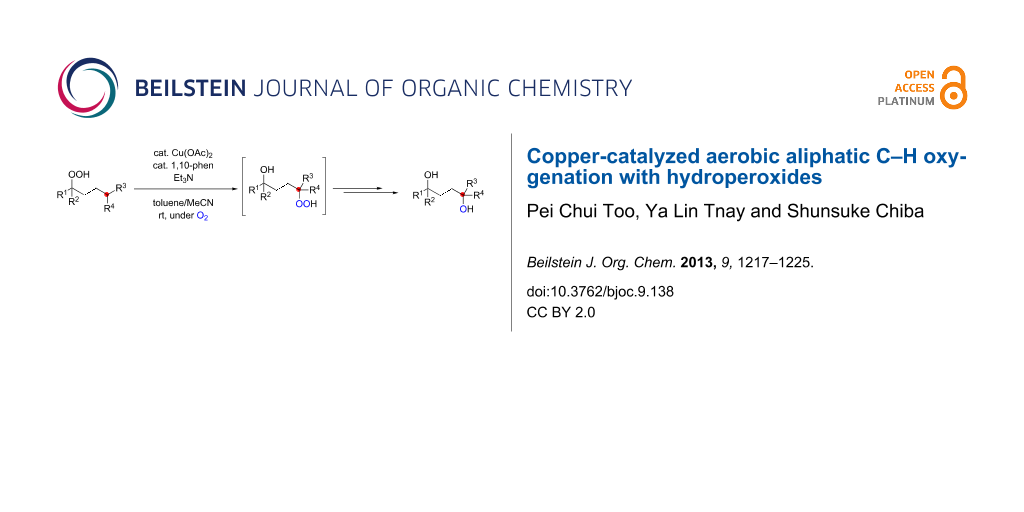
Abstract
We report herein Cu-catalyzed aerobic oxygenation of aliphatic C–H bonds with hydroperoxides, which proceeds by 1,5-H radical shift of putative oxygen-centered radicals (O-radicals) derived from hydroperoxides followed by trapping of the resulting carbon-centered radicals with molecular oxygen.

Graphical Abstract
Introduction
Aliphatic sp3 C–H bonds are ubiquitous components in organic molecules but rather inert towards most of the chemical reactions. It thus remains as one of the most challenging topics in organic synthesis to develop catalytic oxidative sp3 C–H functionalization with predictable chemo- and regioselectivity [1-4]. To achieve this goal, we have recently utilized 1,5-H-radical shift [5,6] with iminyl radical species (N-radicals) generated under Cu-catalyzed aerobic reaction conditions, in which the resulting carbon-centered radicals (C-radicals) could be trapped by molecular oxygen to form new C–O bonds. For instance, the Cu-catalyzed aerobic reaction of N-alkylamidines afforded aminidyl radicals (N-radicals) by single-electron oxidation and deprotonation of the amidine moiety, which was followed by 1,5-H-radical shift to generate the corresponding C-radicals (Scheme 1a) [7]. The successive trapping of the resulting C-radicals with molecular O2 forms peroxy radicals (the C–O bond formation). Reduction of peroxy radicals generates alkoxides, cyclization of which with the amidine moiety finally affords dihydrooxazoles. Similarly, it was also revealed that the sp3 C–H oxygenation could proceed directed by the N–H ketimine moieties under Cu-catalyzed aerobic conditions via the corresponding iminyl radical species, where 1,2-diacylbenzenes and amino endoperoxides could be synthesized by C–H oxygenation of secondary and tertiary C–H bonds, respectively (Scheme 1b) [8,9].
![[1860-5397-9-138-i1]](/bjoc/content/inline/1860-5397-9-138-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Aliphatic C–H oxidation with amidines and ketimines by 1,5-H radical shift.
Scheme 1: Aliphatic C–H oxidation with amidines and ketimines by 1,5-H radical shift.
Stimulated by these remote sp3 C–H oxidation reactions using the nitrogen-centered radicals derived from amidines and ketimines, we became interested to utilize oxygen-centered radicals (O-radicals) for the sp3 C–H functionalization. In this context, we envision employing hydroperoxides as precursors for O-radicals in the presence of Cu salts. The lower valent Cu(I) species could potentially undergo single-electron reduction of hydroperoxides to produce the corresponding O-radicals [10-13]. Ball recently reported CuCl-catalyzed aliphatic C–H chlorination using hydroperoxides as the O-radical source, in which the C-radicals generated by the 1,5-H radical shift were chlorinated (Scheme 2a) [14]. If reductive generation of the O-radicals from hydroperoxides could be achieved under an O2 atmosphere, the C-radicals generated by 1,5-H radical shift could be trapped by O2 to form the new C–O bonds. Herein, we report the realization of this concept mainly for the aerobic synthesis of 1,4-diols from hydroperoxides, which could be catalyzed by the Cu(OAc)2-1,10-phenanthroline system in the presence of Et3N as a terminal reductant of the Cu(II) species (Scheme 2b).
![[1860-5397-9-138-i2]](/bjoc/content/inline/1860-5397-9-138-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Aliphatic C–H oxidation with hydroperoxides.
Scheme 2: Aliphatic C–H oxidation with hydroperoxides.
Findings
We commenced our investigation with the Cu-catalyzed aerobic reactions of hydroperoxide 1a in the presence of Et3N as a terminal reductant [15] to keep lower valent Cu(I) species in the reaction mixture (Table 1). As expected, when 1a was treated with Cu(OAc)2 (20 mol %) in the presence of Et3N (2 equiv) in DMF, the reaction proceeded at room temperature for 17 h to afford C–H oxygenation products, cyclic hemiacetal 2a and 1,4-diol 3a in 49% and 8% yields, respectively (Table 1, entry 1). It was found that addition of nitrogen ligands such as 2,2’-bipyridine and 1,10-phenanthroline (1,10-phen) accelerated the reaction (Table 1, entries 2 and 3). The reactions with CuCl2 (Table 1, entry 4) as well as CuCl (Table 1, entry 5) resulted in comparable results with that by Cu(OAc)2. Further optimization of the reaction conditions by the solvent screening (Table 1, entries 6–11) revealed that the co-solvent system (benzene/MeCN or toluene/MeCN) performed best to give the highest yield (83–87% combined yields of 2a and 3a, Table 1, entries 9–11), in which the amount of Et3N could be reduced to 0.5 equiv (Table 1, entries 10 and 11). It is worthy of note that the catalytic loading of Cu(OAc)2-1,10-phen could be lowered to 5 mol % while maintaining good combined yields of 2a and 3a (89%, Table 1, entry 13). Upon completion of the C–H oxygenation under the reaction conditions in entry 13, the resulting crude residue after removal of the solvents was treated with LiAlH4 in THF, affording 1,4-diol 3a as the sole product in 90% yield (Table 1, entry 14). The Cu-catalyzed reaction of 1a under an inert (N2) atmosphere gave an intramolecular C–H oxygenation product, dihydroisobenzofuran 4a in 47% yield (Table 1, entry 15).
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-138-i8.svg?max-width=637&scale=1.0)
|
||||||||
| Entry |
Cu salts
(mol %) |
Additive
(mol %) |
Et3N
(equiv) |
Solvent
(0.1 M) |
Time
(h) |
Yield (%)b | ||
|---|---|---|---|---|---|---|---|---|
| 2a | 3a | 4a | ||||||
| 1 |
Cu(OAc)2
(20) |
– | 2.0 | DMF | 17 | 49 | 8 | 0 |
| 2 |
Cu(OAc)2
(20) |
2,2’-bipyridine
(20) |
2.0 | DMF | 3 | 49 | 9 | 0 |
| 3 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
2.0 | DMF | 3 | 53 | 12 | 0 |
| 4 |
CuCl2
(20) |
1,10-phen
(20) |
2.0 | DMF | 6 | 52 | 10 | 0 |
| 5 |
CuCl
(20) |
1,10-phen
(20) |
2.0 | DMF | 3 | 52 | 10 | 0 |
| 6 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
2.0 | MeCN | 2 | 55 | 20 | 0 |
| 7 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
2.0 | benzene | 5 | 71 | 13 | 0 |
| 8 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
2.0 | toluene | 9 | 70 | 14 | 0 |
| 9 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
2.0 |
benzene/MeCN
(5:1) |
2 | 76 | 11 | 0 |
| 10 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
0.5 |
benzene/MeCN
(5:1) |
3 | 74 | 9 | 0 |
| 11 |
Cu(OAc)2
(20) |
1,10-phen
(20) |
0.5 |
toluene/MeCN
(5:1) |
3 | 74 | 10 | 0 |
| 12 |
Cu(OAc)2
(10) |
1,10-phen
(10) |
0.5 |
toluene/MeCN
(5:1) |
3 | 75 | 16 | 0 |
| 13 |
Cu(OAc)2
(5) |
1,10-phen
(5) |
0.5 |
toluene/MeCN
(5:1) |
3 | 75 | 14 | 0 |
| 14c |
Cu(OAc)2
(5) |
1,10-phen
(5) |
0.5 |
toluene/MeCN
(5:1) |
3 | – | 90 | 0 |
| 15d |
Cu(OAc)2
(5) |
1,10-phen
(5) |
0.5 |
toluene/MeCN
(5:1) |
1 | 0 | 0 | 47 |
aReactions were carried out using 0.3 mmol of hydroperoxide 1a in solvents (3 mL, 0.1 M) at room temperature under an O2 atmosphere. bIsolated yields are recorded. cAfter stirring 5 h, the volatile materials were removed in vacuo, and the resulting crude materials were further treated with LiAlH4 (1.2 equiv) in THF at rt for 1 h. dThe reaction was conducted under a N2 atmosphere.
The proposed reaction pathways for the formation of 2a, 3a, and 4a were described in Scheme 3. Single-electron reduction of the starting Cu(OAc)2 by Et3N forms the Cu(I) species, which reduces hydroperoxide 1a to give O-radical I along with the generation of the Cu(II) species. 1,5-H-Radical shift of O-radical I generates C-radical II, which is trapped by molecular O2 to give peroxy radical III. Probably further reaction of III with Cu(I) species gives Cu(II)-peroxide IV, which undergoes fragmentation to give aldehyde V [16-18], which in turn cyclizes to afford hemiacetal 2a. Protonation of Cu(II)-peroxide IV followed by the reduction of the resulting hydroperoxide deliver 1,4-diol 3a. In the absence of molecular O2 (under a N2 atmosphere, Table 1, entry 15), the resulting C-radical I is oxidized by the Cu(II) species to give carbocation VI [19], which is trapped by the intramolecular hydroxy group to give 4a.
![[1860-5397-9-138-i3]](/bjoc/content/inline/1860-5397-9-138-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed reaction mechanisms for the formation of 2a, 3a, and 4a.
Scheme 3: Proposed reaction mechanisms for the formation of 2a, 3a, and 4a.
We next explored the substrate scope of the present aerobic strategy for the synthesis of 1,4-diols by targeting methylene C–H oxygenation with various tertiary hydroperoxides 1 (Table 2). Generally, oxygenation of benzylic methylene C–H bonds proceeded smoothly to give the corresponding 1,4-diols 3 in good to moderate yields (77–51% yields) (Table 2, entries 1–7). Moreover, the present method allowed for oxygenation of nonbenzylic methylene C–H bonds, while yields of obtained 1,4-diols 3 were relatively low (65–40% yields) (Table 2, entries 8–13). In most cases (except for Table 2, entries 3, 6, 7 and 11), either reduced alcohols 5 (up to 23% yield) or fragmented alcohols 6 (up to 23% yield), or both were observed as minor products. The formation of alcohols 5 occurs by two-electron reduction of hydroperoxides 1 followed by protonation or by H-radical abstraction of the transient O-radical generated by one-electron reduction of hydroperoxides 1 prior to the 1,5-H radical shift (Scheme 4). Presumably, radical fragmentation of O-radicals occurs to give the corresponding ketones 7, which are reduced by LiAlH4 in the next step to give alcohols 6 (Scheme 4).
Table 2: Substrate scope: oxygenation of secondary C–H bonds.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-138-i9.svg?max-width=637&scale=1.0)
|
||||
| Entry |
Hydroperoxides
1 |
1,4-Diols
3 |
Others | |
|---|---|---|---|---|
| Alcohols 5 | Alcohols 6 | |||
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-138-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-138-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-138-i12.svg?max-width=637&scale=1.0)
|
||
| 1 | 1b (R2 = Ph) | 3b 77% | – | 6b 8%b |
| 2 | 1c (R2 = Me) | 3c 72% (1:1) | – | 6c 8%b |
| 3 | 1d (R2 = Et) | 3d 51% (1:1) | – | – |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-138-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-138-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-138-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-138-i16.svg?max-width=637&scale=1.0)
|
|
| 4 | 1e (R2 = 2-naphthyl) | 3e 62% (1:1) | 5e 9% | 6e 17%b |
| 5 | 1f (R2 = 4-Br-C6H4) | 3f 70% (1:1) | 5f 15% | 6f 15%b |
| 6 | 1g (R2 = CH2CH2Ph) | 3g 51% (1:1) | – | – |
| 7 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-138-i17.svg?max-width=637&scale=1.0)
1h |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-138-i18.svg?max-width=637&scale=1.0)
3h 56% |
– | – |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-138-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-138-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-138-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-138-i22.svg?max-width=637&scale=1.0)
|
|
| 8 | 1i (R2 = Ph) | 3i 65% | 5i 18% | 6b 14%b |
| 9 | 1j (R2 = Me) | 3j 62% (1.27:1) | 5j 23% | 6c 20%b |
| 10 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-138-i23.svg?max-width=637&scale=1.0)
1k |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-138-i24.svg?max-width=637&scale=1.0)
3k 40% (1:1) |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-138-i25.svg?max-width=637&scale=1.0)
5k 16% |
– |
| 11 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-138-i26.svg?max-width=637&scale=1.0)
1l |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-138-i27.svg?max-width=637&scale=1.0)
3l 48% |
– | – |
| 12 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-138-i28.svg?max-width=637&scale=1.0)
1m |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-138-i29.svg?max-width=637&scale=1.0)
3m 45% (1.27:1) |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-138-i30.svg?max-width=637&scale=1.0)
5m 18% |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-138-i31.svg?max-width=637&scale=1.0)
6c 20%b |
| 13 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-138-i32.svg?max-width=637&scale=1.0)
1n |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-138-i33.svg?max-width=637&scale=1.0)
3n 51% (trans/cis = 1.37:1) |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-138-i34.svg?max-width=637&scale=1.0)
5n 13% |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-138-i35.svg?max-width=637&scale=1.0)
6b 23%b |
aReactions were carried out using 0.5 mmol of hydroperoxides 1 with Cu(OAc)2 (5 mol %), 1,10-phen (5 mol %), and Et3N (0.5 equiv) in toluene/MeCN (5:1, 0.1 M) at room temperature under an O2 atmosphere. After stirring for 4–7 h, the volatile materials were removed in vacuo, and the resulting crude materials were further treated with LiAlH4 (1.2 equiv) in THF at rt for 1 h. Isolated yields are recorded. The ratio in parentheses is the diastereomer ratio of the products 3, where available. b 1H NMR yields with 1,1,2,2-tetrachloroethane as an internal standard.
![[1860-5397-9-138-i4]](/bjoc/content/inline/1860-5397-9-138-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed reaction mechanisms for the formation of 5 and 6.
Scheme 4: Proposed reaction mechanisms for the formation of 5 and 6.
The reaction of a secondary hydroperoxide such as 1o, however, afforded C–H oxygenation product 2o (as a keto form) only in 30% yield along with the formation of the corresponding reduced alcohol 5o and ketone 8o in 10% and 43% yields, respectively (Scheme 5).
![[1860-5397-9-138-i5]](/bjoc/content/inline/1860-5397-9-138-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The reaction of secondary hydroperoxide 1o.
Scheme 5: The reaction of secondary hydroperoxide 1o.
Oxygenation of tertiary C–H bonds was also examined under the present aerobic conditions (Table 3). In these cases, the Cu-catalyzed aerobic reactions afford hydroperoxides 9 as an initial aerobic C–H oxygenation product. After stirring for 5–7 h, the reaction mixtures were successively treated with PPh3 (1 equiv) for the reduction of 9 to obtain 1,4-diols 3. Although the desired 1,4-diols 3 were formed with this two-step one-pot procedure, the isolated yields of 3p–3r were moderate (45–52% yields) (Table 3, entries 1–3). The oxygenation of the adamantyl C–H bond was especially sluggish, giving the desired 1,4-diol 3s in only 23% yield (Table 3, entry 4). Similarly, in these reactions, the formation of reduced alcohols 5 (for Table 3, entries 1 and 2) and fragmented ketones 7 (for Table 3, entries 1–4) were observed as minor products.
Table 3: Substrate scope oxygenation of tertiary C–H bonds.a
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-138-i36.svg?max-width=637&scale=1.0)
|
||||
| Entry |
Hydroperoxides
1 |
1,4-Diols
3 |
Others | |
|---|---|---|---|---|
| Alcohols 5 | Ketones 7 | |||
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-138-i37.svg?max-width=637&scale=1.0)
|
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-138-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-138-i39.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-138-i40.svg?max-width=637&scale=1.0)
|
|
| 1 | 1p (R2 = Ph) | 3p 52% | 5p 16% | 7b 28%b |
| 2 | 1q (R2 = Me) | 3q 45% | 5q 23% | 7c 24%b |
| 3 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-138-i41.svg?max-width=637&scale=1.0)
1r |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-138-i42.svg?max-width=637&scale=1.0)
3r 47% |
– |
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-138-i43.svg?max-width=637&scale=1.0)
7b 12%b |
| 4 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-9-138-i44.svg?max-width=637&scale=1.0)
1s |
![[Graphic 38]](/bjoc/content/inline/1860-5397-9-138-i45.svg?max-width=637&scale=1.0)
3s 23% |
– |
![[Graphic 39]](/bjoc/content/inline/1860-5397-9-138-i46.svg?max-width=637&scale=1.0)
7b 12%b |
aReactions were carried out by using 0.5 mmol of hydroperoxides 1 with Cu(OAc)2 (5 mol %), 1,10-phen (5 mol %), and Et3N (0.5 equiv) in toluene/MeCN (5:1, 0.1 M) at room temperature under an O2 atmosphere. After stirring for 5–7 h, the reaction mixture was further treated with PPh3 (1 equiv) at rt. Isolated yields are recorded. b 1H NMR yields with 1,1,2,2-tetrachloroethane as an internal standard.
Our next challenge was to develop direct aerobic dioxygenation of alkanes using the present radical strategy. The generation of C-radicals from alkanes at C–H bonds bearing relatively weak bond-dissociation enthalpies (i.e., tertiary alkyl C–H bonds, benzylic C–H bonds, etc.), by phthalimide N-oxyl radicals generated oxidatively from N-hydroxyphthalimide (NHPI), has been reported [20-22]. The resulting C-radicals could be trapped with molecular oxygen to form hydroperoxides. It could be envisioned that this aerobic C–H bond oxygenation could be combined with the present remote C–H oxygenation with hydroperoxides, presumably resulting in direct formation of 1,4-dioxygenated compounds from nonoxygenated alkanes (Scheme 6).
![[1860-5397-9-138-i6]](/bjoc/content/inline/1860-5397-9-138-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
With this hypothesis, alkane 10 bearing a dibenzylic tertiary C–H bond (marked in green) was treated with the catalytic system of CuCl-1,10-phen (20 mol %) with NHPI in benzene/MeCN solvent under an O2 atmosphere (1 atm) (Scheme 7). The reaction with 20 mol % of NHPI proceeded as expected at 50 °C to afford a mixture of lactol 2i and 1,4-diols 3i in 29% and 3% yields, respectively, via the desired 1,4-dioxygenation, while benzophenone (7b) was also formed in 30% yield through fragmentation of the transient alkoxy radical (Scheme 4). Use of 40 mol % of NHPI slightly improved the yields of 1,4-dioxygenation products 2i and 3i (40% and 4% yields, respectively).
![[1860-5397-9-138-i7]](/bjoc/content/inline/1860-5397-9-138-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Aerobic 1,4-dioxygenation of alkanes in the CuCl–NHPI catalytic system.
Scheme 7: Aerobic 1,4-dioxygenation of alkanes in the CuCl–NHPI catalytic system.
In conclusion, we have developed the Cu-catalyzed aerobic oxygenation of aliphatic C–H bonds using hydroperoxides by a 1,5-H radical shift of putative O-radicals derived from hydroperoxides followed by trapping of the resulting C-radicals with molecular oxygen. A preliminary result involving the direct 1,4-dioxygenation of alkane 10 was demonstrated by using the present method. Further studies will be carried out to develop more robust and efficient catalytic aerobic radical transformations for polyol synthesis from rather simple alkanes.
Supporting Information
| Supporting Information File 1: Full experimental details and analytical data. | ||
| Format: PDF | Size: 4.7 MB | Download |
References
-
White, M. C. Science 2012, 335, 807–809. doi:10.1126/science.1207661
Return to citation in text: [1] -
Davies, H. M. L.; Du Bois, J.; Yu, J.-Q. Chem. Soc. Rev. 2011, 40, 1855–1856. doi:10.1039/c1cs90010b
Return to citation in text: [1] -
Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. doi:10.1002/anie.201006368
Return to citation in text: [1] -
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e
Return to citation in text: [1] -
Ĉeković, Ž. Tetrahedron 2003, 59, 8073–8090. doi:10.1016/S0040-4020(03)01202-X
Return to citation in text: [1] -
Majetich, G.; Wheless, K. Tetrahedron 1995, 51, 7095–7129. doi:10.1016/0040-4020(95)00406-X
Return to citation in text: [1] -
Wang, Y.-F.; Chen, H.; Zhu, X.; Chiba, S. J. Am. Chem. Soc. 2012, 134, 11980–11983. doi:10.1021/ja305833a
Return to citation in text: [1] -
Zhang, L.; Ang, G. Y.; Chiba, S. Org. Lett. 2011, 13, 1622–1625. doi:10.1021/ol200425c
Return to citation in text: [1] -
Zhu, P.; Tong, B. M. K.; Wang, R.; Chen, J. P.; Foo, S.; Chong, H. C.; Wang, X. L.; Ang, G. Y.; Chiba, S.; Tan, N. S. Cell Death Dis. 2013, 4, e552. doi:10.1038/cddis.2013.68
Return to citation in text: [1] -
Ĉeković, Ž.; Ilijev, D. Tetrahedron Lett. 1988, 29, 1441–1444. doi:10.1016/S0040-4039(00)80319-6
Return to citation in text: [1] -
Ĉeković, Ž.; Ĉvetković, M. Tetrahedron Lett. 1982, 23, 3791–3794. doi:10.1016/S0040-4039(00)87708-4
Return to citation in text: [1] -
Ĉeković, Ž.; Dimitrijević, L.; Djokić, G.; Srnić, T. Tetrahedron 1979, 35, 2021–2026. doi:10.1016/S0040-4020(01)88972-9
Return to citation in text: [1] -
Ĉeković, Ž.; Green, M. M. J. Am. Chem. Soc. 1974, 96, 3000–3002. doi:10.1021/ja00816a059
Return to citation in text: [1] -
Kundu, R.; Ball, Z. T. Org. Lett. 2010, 12, 2460–2463. doi:10.1021/ol100472t
Return to citation in text: [1] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n
Return to citation in text: [1] -
Zhang, L.; Lee, J.-Y.; Yamazaki, N.; Chiba, S. Synlett 2011, 2167–2170. doi:10.1055/s-0030-1261185
Return to citation in text: [1] -
Tnay, Y. L.; Chen, C.; Chua, Y. Y.; Zhang, L.; Chiba, S. Org. Lett. 2012, 14, 3550–3553. doi:10.1021/ol301583y
Return to citation in text: [1] -
Chiba, S.; Zhang, L.; Lee, J.-Y. J. Am. Chem. Soc. 2010, 132, 7266–7267. doi:10.1021/ja1027327
Return to citation in text: [1] -
Kochi, J. K.; Bemis, A.; Jenkins, C. L. J. Am. Chem. Soc. 1968, 90, 4616–4625. doi:10.1021/ja01019a018
Return to citation in text: [1] -
Ishii, Y.; Sakaguchi, S.; Iwahama, T. Adv. Synth. Catal. 2001, 343, 393–427. doi:10.1002/1615-4169(200107)343:5<393::AID-ADSC393>3.0.CO;2-K
Return to citation in text: [1] -
Roduner, E.; Kaim, W.; Sarjar, B.; Urlacher, V. B.; Pleiss, J.; Gläser, R.; Einicke, W.-D.; Sprenger, G. A.; Beifuß, U.; Klemm, E.; Liebner, C.; Hieronymus, H.; Hsu, S.-F.; Plietker, B.; Laschat, S. ChemCatChem 2013, 5, 82–112. doi:10.1002/cctc.201200266
Return to citation in text: [1] -
Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k
Return to citation in text: [1]
| 1. | White, M. C. Science 2012, 335, 807–809. doi:10.1126/science.1207661 |
| 2. | Davies, H. M. L.; Du Bois, J.; Yu, J.-Q. Chem. Soc. Rev. 2011, 40, 1855–1856. doi:10.1039/c1cs90010b |
| 3. | Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. doi:10.1002/anie.201006368 |
| 4. | Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e |
| 10. | Ĉeković, Ž.; Ilijev, D. Tetrahedron Lett. 1988, 29, 1441–1444. doi:10.1016/S0040-4039(00)80319-6 |
| 11. | Ĉeković, Ž.; Ĉvetković, M. Tetrahedron Lett. 1982, 23, 3791–3794. doi:10.1016/S0040-4039(00)87708-4 |
| 12. | Ĉeković, Ž.; Dimitrijević, L.; Djokić, G.; Srnić, T. Tetrahedron 1979, 35, 2021–2026. doi:10.1016/S0040-4020(01)88972-9 |
| 13. | Ĉeković, Ž.; Green, M. M. J. Am. Chem. Soc. 1974, 96, 3000–3002. doi:10.1021/ja00816a059 |
| 8. | Zhang, L.; Ang, G. Y.; Chiba, S. Org. Lett. 2011, 13, 1622–1625. doi:10.1021/ol200425c |
| 9. | Zhu, P.; Tong, B. M. K.; Wang, R.; Chen, J. P.; Foo, S.; Chong, H. C.; Wang, X. L.; Ang, G. Y.; Chiba, S.; Tan, N. S. Cell Death Dis. 2013, 4, e552. doi:10.1038/cddis.2013.68 |
| 7. | Wang, Y.-F.; Chen, H.; Zhu, X.; Chiba, S. J. Am. Chem. Soc. 2012, 134, 11980–11983. doi:10.1021/ja305833a |
| 5. | Ĉeković, Ž. Tetrahedron 2003, 59, 8073–8090. doi:10.1016/S0040-4020(03)01202-X |
| 6. | Majetich, G.; Wheless, K. Tetrahedron 1995, 51, 7095–7129. doi:10.1016/0040-4020(95)00406-X |
| 19. | Kochi, J. K.; Bemis, A.; Jenkins, C. L. J. Am. Chem. Soc. 1968, 90, 4616–4625. doi:10.1021/ja01019a018 |
| 16. | Zhang, L.; Lee, J.-Y.; Yamazaki, N.; Chiba, S. Synlett 2011, 2167–2170. doi:10.1055/s-0030-1261185 |
| 17. | Tnay, Y. L.; Chen, C.; Chua, Y. Y.; Zhang, L.; Chiba, S. Org. Lett. 2012, 14, 3550–3553. doi:10.1021/ol301583y |
| 18. | Chiba, S.; Zhang, L.; Lee, J.-Y. J. Am. Chem. Soc. 2010, 132, 7266–7267. doi:10.1021/ja1027327 |
| 20. | Ishii, Y.; Sakaguchi, S.; Iwahama, T. Adv. Synth. Catal. 2001, 343, 393–427. doi:10.1002/1615-4169(200107)343:5<393::AID-ADSC393>3.0.CO;2-K |
| 21. | Roduner, E.; Kaim, W.; Sarjar, B.; Urlacher, V. B.; Pleiss, J.; Gläser, R.; Einicke, W.-D.; Sprenger, G. A.; Beifuß, U.; Klemm, E.; Liebner, C.; Hieronymus, H.; Hsu, S.-F.; Plietker, B.; Laschat, S. ChemCatChem 2013, 5, 82–112. doi:10.1002/cctc.201200266 |
| 22. | Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k |
© 2013 Too et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)