Abstract
Since the beginning of the century, N-hydroxyphthalimide and related compounds have been revealed to be efficient organocatalysts for free-radical processes and have found ample application in promoting the aerobic oxidation of a wide range of organic substrates. When combined with different co-catalysts, they are activated to the corresponding N-oxyl radical species and become able to promote radical chains, involving molecular oxygen, directly or indirectly. Most of the examples reported in the literature describe the use of these N-hydroxy derivatives in the presence of transition-metal complexes. However, eco-friendly standards, including the demand for highly selective transformations, impose the development of metal-free processes, especially for large-scale productions, as in the case of the oxygenation of hydrocarbons. For this reason, many efforts have been devoted in the past decade to the design of new protocols for the activation of N-hydroxy imides in the presence of nonmetal initiators. Herein we provide a concise overview of the most significant and successful examples in this field, with the final aim to furnish a useful instrument for all scientists actively involved in the O2-mediated selective oxidation of organic compounds and looking for environmentally safe alternatives to metal catalysis.
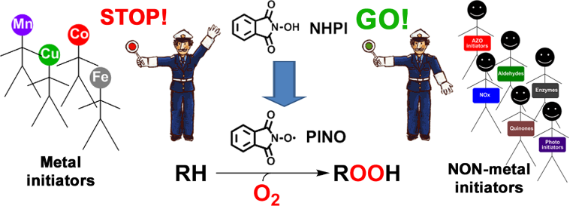
Graphical Abstract
Introduction
The development of efficient and cheap catalytic systems for the selective oxidation of organic substrates under mild and environmentally benign conditions represents one of the major challenges in organic synthesis [1]. In this context, the replacement of traditional oxidants, often used in stoichiometric amounts, with molecular oxygen is mandatory in order to improve the beneficial impact of selective oxidation on industrial chemistry [2-5]. Nevertheless, classical autoxidation is usually very slow at low temperatures, and catalysis is required to activate O2. Transition-metal salts are particularly effective for this scope [6], but their use is often detrimental for the selectivity of the process and they would not meet the standards of “green chemistry”. An alternative catalytic route is based on the use of N-hydroxy imides (NHIs), and in particular N-hydroxyphthalimide (NHPI), which have found ample application as ideal catalysts for the aerobic oxidation of organic substrates [7-11].
NHPI acts as a precursor of the phthalimide N-oxyl (PINO) radical, which is the effective catalyst promoting hydrogen-abstraction processes (Scheme 1).
![[1860-5397-9-146-i1]](/bjoc/content/inline/1860-5397-9-146-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Catalytic role of NHPI in the selective oxidation of organic substrates.
Scheme 1: Catalytic role of NHPI in the selective oxidation of organic substrates.
The reactivity of NHPI and PINO is related to the bond dissociation energy (BDE) of the O–H group, which was estimated at 88.1 kcal/mol [12]. This value is similar to the BDE of O–H in hydroperoxides, suggesting that the faster reactivity of PINO compared to peroxyl radicals should be attributed to an enhanced polar effect involved in the hydrogen abstraction by this nitroxyl radical [13]. Furthermore, NHPI also behaves as a relatively good hydrogen donor even at low temperatures (kH = 7.2 × 103 M−1s−1) [12], trapping peroxyl radicals before they undergo termination.
PINO generation represents the key step of the overall process. Many transition metal salts and complexes have been successfully used as co-catalysts for NHPI activation. However, once again their use should be avoided in order to improve the sustainability of the process. For this reason, in the past decade several efforts have been devoted to the development of catalytic systems for the metal-free activation of NHIs under mild conditions. With an overview on the results reported in the literature in the past decade, we aim to describe herein the most significant examples related to the selective oxidation of organic molecules with molecular oxygen, catalyzed by NHIs in the presence of nonmetal cocatalysts. After briefly describing the role of classical radical initiators obtained by thermal decomposition, we will focus on some intriguing redox systems, including nitric oxides, laccase, quinones and aldehydes, which allow operation under very mild conditions, offering efficient alternative solutions to the classical autoxidation processes, especially in the field of the selective oxygenation of hydrocarbons.
Review
Radical initiation by thermal decomposition
Thermal decomposition of peroxides and azo-compounds is a well-known technique generally used to generate radicals in solution. Ishii and co-workers widely investigated the key role of these initiators in promoting the formation of PINO, performing several selective transformations under aerobic or anaerobic conditions.
The combination of NHPI with tiny amounts of dibenzoyl peroxide (BPO) under an atmosphere of argon led to the hydroacylation of simple alkenes by addition of acyl radicals derived from aldehydes and masked aldehydes, such as 1,3-dioxolanes (Scheme 2) [14,15].
![[1860-5397-9-146-i2]](/bjoc/content/inline/1860-5397-9-146-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Radical addition of aldehydes and analogues to alkenes.
Scheme 2: Radical addition of aldehydes and analogues to alkenes.
The mediating role of NHPI allowed this approach to be extended to electron-deficient olefins [15], preventing the polymerization that easily occurs with these substrates in the presence of radical initiators.
Azo-initiators were mainly employed for the synthesis of phenol derivatives by aerobic oxidation of isopropyl aromatics. For example, the oxidation of 2,6-diisopropylnaphthalene with air in the presence of α,α’-azobisisobutyronitrile (AIBN) and NHPI, followed by decomposition with sulfuric acid of the dihydroperoxide oxidation product, gave the corresponding 2,6-naphthalenediol in 92% yield (Scheme 3) [16].
![[1860-5397-9-146-i3]](/bjoc/content/inline/1860-5397-9-146-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: NHPI/AIBN-promoted aerobic oxidation of 2,6-diisopropylnaphthalene.
Scheme 3: NHPI/AIBN-promoted aerobic oxidation of 2,6-diisopropylnaphthalene.
As expected, the replacement of AIBN with a transition-metal complex (Co(OAc)2) resulted in a collapse of selectivity due to the redox decomposition of the hydroperoxides. This catalytic system was also applied to the oxygenation of 1,3,5-triisopropylbenzene [17]. However, in this case the conversion of all isopropyl groups was far from being reached, with mono- and di-phenols being the major products, while the yield in benzene-1,3,5-triol was close to 1%.
Sheldon and co-workers tested several commercially available azo-compounds (including AIBN and 2,2’-azobis(2,4-dimethylvaleronitrile) (AMVN)) as radical initiators at different temperatures for the NHPI-catalyzed oxidation of cyclohexylbenzene (CHB) to the corresponding hydroperoxides, finding a high selectivity in the oxygenation at the 1-position (CHBHP, Scheme 4) [18]. The selectivity decreased rapidly with increasing conversion and temperature. The use of CHBHP itself as radical initiator in place of azo-compounds required higher temperatures (100–120 °C), leading to conversions up to 20%, but to the detriment of the selectivity (72%).
![[1860-5397-9-146-i4]](/bjoc/content/inline/1860-5397-9-146-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: NHPI/AIBN-promoted aerobic oxidation of CHB.
Scheme 4: NHPI/AIBN-promoted aerobic oxidation of CHB.
An analogous catalytic system was employed by Punta et al. [19] for the selective peroxidation of polyunsaturated fatty acids (PUFA), a transformation of high biological interest [20]. The combination of N-methylbenzohydroxamic acid (NMBHA) with 2,2’-azobis(4-methoxy-2,4-dimethylvaleronitrile) (MeO-AMVN), an azo initiator that decomposes at physiological temperatures 15 times faster than the corresponding AMVN, allowed the trans,cis conjugated hydroperoxides to be obtained exclusively and in good yields by direct oxygenation of the corresponding lipids. In fact, due to its ideal O–H BDE value (79.2 kcal/mol [12]), NMBHA turned out to be a better candidate than NHPI to act both as autoxidation catalyst, promoting the hydrogen abstraction from the bisallylic C–H position of the fatty esters by means of the corresponding amidoxyl radical, and as a good hydrogen donor (kH = 1.2 × 105 M−1s−1 [19]) trapping trans,cis peroxyl radicals before they underwent β-fragmentation (Scheme 5).
![[1860-5397-9-146-i5]](/bjoc/content/inline/1860-5397-9-146-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: NMBHA/MeOAMVN promoted aerobic oxidation of PUFA.
Scheme 5: NMBHA/MeOAMVN promoted aerobic oxidation of PUFA.
In accordance with this free-radical mechanism, Schmidt and Alexanian more recently reported the aerobic dioxygenation of alkenyl N-aryl hydroxamic acids (Scheme 6) [21]. In this case dioxygenation proceeded under 1 atmosphere of O2 alone, without any additional initiator being required.
![[1860-5397-9-146-i6]](/bjoc/content/inline/1860-5397-9-146-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Alkene dioxygenation by means of N-aryl hydroxamic acid and O2.
Scheme 6: Alkene dioxygenation by means of N-aryl hydroxamic acid and O2.
Radical initiation by redox processes
Nitrogen oxides and nitric acid
The PINO activation by means of nitric oxide was first reported by Ishii et al. in 1997 [22]. By reacting adamantane in a mixed solvent of benzonitrile and acetic acid under an atmosphere of NO and in the presence of catalytic amounts of NHPI he observed the formation of 1-N-adamantylbenzamide as a principal product (Scheme 7), while when operating under the same conditions but in the presence of molecular oxygen, 1-nitroadamantane was achieved in good yields.
![[1860-5397-9-146-i7]](/bjoc/content/inline/1860-5397-9-146-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: NHPI-catalyzed reaction of adamantane under NO atmosphere.
Scheme 7: NHPI-catalyzed reaction of adamantane under NO atmosphere.
Moreover, by simply moving into acetonitrile, they observed the selective oxygenation of phthalane to the corresponding phthalaldehyde in 80% yield [23]. In both cases the reaction proceeds via the formation of a carbocation intermediate (Scheme 7).
The same research group also reported the efficient air-assisted nitration of alkanes [24] and alkyl side-chain aromatic compounds [25] by nitrogen dioxide and nitric acid, under NHPI catalysis (Scheme 8).
![[1860-5397-9-146-i8]](/bjoc/content/inline/1860-5397-9-146-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Nitration of alkanes and alkyl side-chains of aromatics.
Scheme 8: Nitration of alkanes and alkyl side-chains of aromatics.
Both HNO3 and NO2 are able to promote the formation of PINO according to path (a) and (b) reported in Scheme 9. These initiation steps lead to the formation of HNO2 which, in turn, is converted into HNO3, H2O, and NO. The latter is oxidized by molecular oxygen back to NO2, thus justifying the higher efficiency of the NO2/air system if compared with anaerobic conditions.
![[1860-5397-9-146-i9]](/bjoc/content/inline/1860-5397-9-146-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Radical mechanism for the nitration of alkanes catalyzed by NHPI.
Scheme 9: Radical mechanism for the nitration of alkanes catalyzed by NHPI.
In this context, we reported in 2004 that the HNO3/O2/I2 system could promote the nitric aerobic oxidation of alkylbenzenes under NHPI-catalysis, leading to the selective formation of benzyl alcohols through the corresponding acetates, if operating in acetic acid solution (Scheme 10) [26]. According to the proposed mechanism, being that the concentrations of NO2 and O2 are much lower than that of I2, benzyl radicals generated from hydrogen abstraction by PINO react faster with the latter one, forming benzyl iodides selectively. Under the described reaction medium benzyl iodides undergo solvolysis, affording the corresponding benzyl acetates in excellent yields.
![[1860-5397-9-146-i10]](/bjoc/content/inline/1860-5397-9-146-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Benzyl alcohols from alkylbenzenes.
Scheme 10: Benzyl alcohols from alkylbenzenes.
Enzyme laccase
Enzyme laccase is a family of “blue-copper” oxidase proteins, containing four copper ions in the active site, which cooperates in the degradation of the biopolymer lignin in woody tissues. With respect to other powerful oxidant enzymes, laccase has a lower redox potential. For this reason, its catalytic action by promoting single-electron oxidation steps is effective only on the phenolic moieties of lignin, with the concomitant reduction of oxygen to water.
Nevertheless, the use of suitable mediators allowed the field of application of this enzyme to be extended to the catalytic oxidation of a wider range of nonphenolic substrates. Among the huge number of mediators, N-hydroxy derivatives (NHDs) turned out to be particularly valuable and have been widely investigated. The role of NHDs-mediators in the laccase oxidation is outlined in Scheme 11. They act as electron carriers that, once oxidized by the enzyme, diffuse in the reaction medium and in turn oxidize those organic substrates unable to enter the enzymatic pocket due to their size. Even if the activity of laccase should be ascribed to the Cu(I) inner core, the enzymatic nature of this oxidizing system allows us to consider this approach as an example of the metal-free activation of NHIs, as no further addition of transition-metal complexes is required.
![[1860-5397-9-146-i11]](/bjoc/content/inline/1860-5397-9-146-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Catalytic cycle of laccase-NHDs mediator oxidizing system.
Scheme 11: Catalytic cycle of laccase-NHDs mediator oxidizing system.
Galli and co-workers [27,28] significantly contributed over the years to the interpretation of the reaction mechanism by comparing NHPI and many other NHDs (Figure 1) with 2,2’-azinobis(3-ethylbenzthiazoline-6-sulfonate) (ABTS), which was the first to be used among mediators of laccase [29]. Laccase-NHDs mediator systems were successfully employed for the aerobic oxidation of nonphenolic substrates such as benzyl alcohols [27,28,30,31] and ethers [32].
![[1860-5397-9-146-1]](/bjoc/content/figures/1860-5397-9-146-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Whereas laccase-ABTS follows an electron transfer (ET) mechanism, NHPI, VLA, HBT, and NHAmediators promote a hydrogen atom transfer (HAT) route through the formation of the corresponding N-oxyl radicals as NHDs-Medox species (Scheme 11). The same research group also emphasized the specialization of mediators versus the substrate. In fact, the laccase-TEMPO system, which operates through an ionic route by formation of oxammonium ion as NHDs-Medox, resulted in particularly efficient promotion of the oxidation of benzyl alcohols, while it gave poor performances when applied in the presence of ethers. In contrast, laccase-NHPI and laccase-HBT systems, which follow a radical mechanism, showed high catalytic activity for the oxidation of ethers.
Quinones and analogous derivatives
As stressed before, the promotion of biological oxygenation is usually mediated by one-electron transfers, which lead to the formation of radicals. On the basis of this consideration, Xu and co-workers suggested that quinones, ubiquitous in nature and often involved in ET chains, could be employed to design biomimetic oxygenation models for the activation of NHPI [33]. The catalytic redox cycle is reported in Scheme 12.
![[1860-5397-9-146-i12]](/bjoc/content/inline/1860-5397-9-146-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: DADCAQ/NHPI-mediated aerobic oxidation mechanism.
Scheme 12: DADCAQ/NHPI-mediated aerobic oxidation mechanism.
The one-electron-transfer interaction of antraquinone (AQ) with NHPI in zeolite HY, followed by hydrogen-atom transfer, successfully led to the formation of the PINO radical, which in turn was responsible for the propagation of the radical chain in the selective oxidation of ethylbenzene to the corresponding acetophenone [33]. Among the different AQ derivatives that were tested, 1,4-diamino-2,3-dichloro-antraquinone (DADCAQ) was the most effective in terms of both conversion and selectivity of the ketone product.
The potential of this catalytic system was proved by extending its application, in the absence of zeolite, for the oxygenation of a wider range of hydrocarbons [34].
Moreover, the electronic effect of substituents on quinones and on the aromatic ring of NHPI was also investigated.
Quinones bearing halogen groups were used in the selective oxidation of alkylarenes, alkenes and alkanes [35], revealing how the moderate electron-withdrawing power of the substituents had a beneficial effect on the ET process. The combined activity of tetrabromo-1,4-benzoquininone (TBBQ) and NHPI afforded the best results.
Aryl-tetrahalogenated NHPI derivatives were also prepared and used in combination with DADCAQ for the oxidation of ethylbenzene [36]. In particular, aryl-tetrachloro-NHPI (TCNHPI) allowed significantly higher conversion and selectivity with respect to the NHPI/DADCAQ classical system (Scheme 13).
![[1860-5397-9-146-i13]](/bjoc/content/inline/1860-5397-9-146-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: DADCAQ/TCNHPI mediated aerobic oxidation of ethylbenzene.
Scheme 13: DADCAQ/TCNHPI mediated aerobic oxidation of ethylbenzene.
More recently, analogous results were achieved by combining NHPI with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) [37].
Xu et al. also reported a similar one-electron-transfer activation of NHPI promoted by nonmetal xanthone and tetramethylammonium chloride (TMAC), for the selective oxidation of hydrocarbons (Scheme 14) [38]. In the proposed mechanism, TMAC has the unique role of decomposing the hydroperoxide intermediate, prolonging the free-radical chain [39-41].
![[1860-5397-9-146-i14]](/bjoc/content/inline/1860-5397-9-146-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: NHPI/xanthone/TMAC mediated aerobic oxidation of ethylbenzene.
Scheme 14: NHPI/xanthone/TMAC mediated aerobic oxidation of ethylbenzene.
The NHPI-activation by AQ has been also adopted by other research groups. Li and co-workers applied the NHPI/AQ system to promote the metal and solvent-free oxidation of α-isophorone to ketoisophorone, preventing the isomerization process of the substrate to β-isophorone (Scheme 15) [42,43].
![[1860-5397-9-146-i15]](/bjoc/content/inline/1860-5397-9-146-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: NHPI/AQ-mediated aerobic oxidation of α-isophorone.
Scheme 15: NHPI/AQ-mediated aerobic oxidation of α-isophorone.
Very recently, Coseri et al. reported that NHPI [44] and other nonpersistent nitroxyl radical precursors, such as VLA, HBT and N-hydroxy-3,4,5,6-tetraphenylphthalimide (TPNHPI) [45], were suitable catalysts for the selective oxidation of cellulose fibers promoted by the NaClO/NaBr system (Scheme 16). According to the proposed mechanism PINO radical is oxidized to the corresponding N-oxammonium cation, which in turn is responsible for the oxidation of the C6 alcoholic function.
![[1860-5397-9-146-i16]](/bjoc/content/inline/1860-5397-9-146-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: NHPI/AQ-mediated oxidation of cellulose fibers by NaClO/NaBr system.
Scheme 16: NHPI/AQ-mediated oxidation of cellulose fibers by NaClO/NaBr system.
The surface modification of cellulose fibers by selectively converting primary hydroxyl groups to the corresponding carboxylic functions, maintaining the original backbone of the polysaccharide, is of major interest for different applications [46,47].
By comparing different activation approaches, including transition-metal complexes, Coseri found that the NHPI/AQ catalytic system allowed higher conversion of hydroxyl groups. A comparison study on the effect of TEMPO and PINO radicals on the oxidation efficiency toward cellulose led to the conclusion that the NHPI/AQ oxidation mediator affords the highest content of carboxylic groups and better preserves the morphology and the molecular weight of the starting material [48].
Moreover, this catalytic system could be employed with dioxygen in place of NaClO as the ultimate oxidizing agent [49]. In this case, the mechanism follows a radical chain via classical HAT by PINO abstraction (Scheme 17).
![[1860-5397-9-146-i17]](/bjoc/content/inline/1860-5397-9-146-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: NHPI/AQ mediated aerobic oxidation of cellulose fibers.
Scheme 17: NHPI/AQ mediated aerobic oxidation of cellulose fibers.
Aldehydes and the molecule-induced homolysis
Sacrificial reductants have been widely reported in the literature as reactive agents able to promote the autoxidation reaction of less-reactive hydrocarbons. In this context, aldehydes have attracted increasing attention [50].
In principle, the aerobic co-oxidation promoted by aldehydes could be considered a nongreen and expensive process, due to the need for sacrificial reagents. However, this approach for oxygen activation could become competitive for practical application if the high efficiency in substrate conversion, the high selectivity in the final product, and/or the field of application, together with the low cost and environmental impact of the selected aldehyde, were significant enough to justify the reagent sacrifice. Moreover, co-oxidation of aldehydes would become attractive also if the acyl derivatives were not used as stoichiometric reagents, but just in catalytic amounts as initiators of free-radical chains.
The use of aldehydes for the activation of NHPI in an aerobic co-oxidative process was first reported by Einhorn and co-workers in 1997 [51]. The combination of stoichiometric amounts of acetaldehyde with catalytic quantities of NHPI promoted the oxidation of a wide range of hydrocarbons, including cumene and ethylbenzene, to the corresponding carbonyl groups. Under these operative conditions, peracetic acid was directly responsible for the substrate oxidation.
More recently we suggested that the NHPI/aldehyde system could promote the formation of PINO radical following a molecule-induced homolysis mechanism [52]. Molecule-induced initiation is a process driven by a thermodynamic effect and consists of a bimolecular reaction according to which an OH radical, generated from hydroperoxides or peracids, undergoes hydrogen abstraction from a suitable molecule bearing relatively weak X–H bonds. The result is the formation of two radical species and a molecule of water (Scheme 18) [53-55].
![[1860-5397-9-146-i18]](/bjoc/content/inline/1860-5397-9-146-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Molecule-induced homolysis by peracids.
Scheme 18: Molecule-induced homolysis by peracids.
We assumed that an analogous homolysis induced by peracids could occur for this NHPI, leading to the formation of PINO radical under mild and metal-free conditions.
This hypothesis was supported by spectroscopic and analytical evidence. By simply adding NHPI to a solution of acetonitrile containing m- chloroperbenzoic acid we could observe the characteristic Electron Paramagnetic Resonance (EPR) spectrum of PINO radical, while m-chlorobenzoic acid (90%) and Cl-benzene (10%) resulted as the unique reaction products (Scheme 19, path a) [52]. When the analogous experiment was conducted in benzene as solvent, m-chlorobenzoate and m-chlorobiphenyl were also detected (Scheme 19, path b).
![[1860-5397-9-146-i19]](/bjoc/content/inline/1860-5397-9-146-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Molecule-induced homolysis of NHPI/m- chloroperbenzoic acid system.
Scheme 19: Molecule-induced homolysis of NHPI/m- chloroperbenzoic acid system.
On the basis of these experimental results, we suggested that the aerobic oxidation of aldehydes could be performed for the in situ generation of the corresponding peracids in the presence of NHPI, promoting co-oxidative processes catalyzed by PINO.
In an early protocol, we reported the NHPI-catalyzed selective aerobic epoxidation of α-olefins and cyclic olefins in the presence of stoichiometric amounts of aldehydes [52].
The experimental results revealed an opposite selectivity with respect to classical epoxidation by peracids, with internal olefins, which were unreactive under our operating conditions. We suggested a free-radical mechanism according to which the acyl peroxyl radical generated in situ is the real epoxidizing agent (Scheme 20).
![[1860-5397-9-146-i20]](/bjoc/content/inline/1860-5397-9-146-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Proposed mechanism for the NHPI/CH3CHO/O2-mediated epoxidation.
Scheme 20: Proposed mechanism for the NHPI/CH3CHO/O2-mediated epoxidation.
This protocol was successfully applied on a larger scale (1 liter Büchi glass vessel) for the synthesis of propylene oxide from the corresponding propene [56], and more recently under continuous-flow conditions, by means of a new multijet oscillating disk (MJOD) reactor, designed and developed by Bjørsvik and co-workers [57]. In this latter case we succeeded in accelerating the overall process, shortening the residence time with respect to the batch protocol.
On the basis of this mechanistic evidence, we also decided to investigate the Einhorn’s process for the oxidation of cumene and ethylbenzene more thoroughly, as we were expected to find a high selectivity in the corresponding hydroperoxides, which were not mentioned in the first report.
Moreover, we also assumed that high amounts of aldehyde were detrimental for the selectivity of the process, the aldehyde having the unique role of initiating the radical chain by generating PINO radical by molecule-induced homolysis.
Indeed, our hypotheses were confirmed and we succeeded in increasing the selectivity in hydroperoxides up to values higher than 80% by simply reducing the amount of acetaldehyde to 10% mol ratio with respect to the alkyl aromatic [58,59] (Scheme 21).
![[1860-5397-9-146-i21]](/bjoc/content/inline/1860-5397-9-146-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: NHPI/CH3CHO-mediated aerobic oxidation of alkyl aromatics.
Scheme 21: NHPI/CH3CHO-mediated aerobic oxidation of alkyl aromatics.
Thus, while the NHPI/AQ catalytic system was particularly effective in converting alkylaromatics to the corresponding carbonyl derivatives, this approach represents a valuable alternative when hydroperoxides are the desired products.
Even if the presence of a polar solvent is crucial to maintain the polar catalyst in solution, it was possible to conduct the aerobic oxidation of cumene at 70 °C in the presence of 1% NHPI, 2% of acetaldehyde, and with a volume ratio cumene/CH3CN of 5/2, achieving the desired hydroperoxide in 28% yield with 84% of selectivity. Similarly, ethylbenzene was oxidized to the corresponding hydroperoxide with a lower yield (13%), but a higher selectivity (91%), by operating at the same temperature with 2% NHPI, 2% acetaldehyde, and with a volume ratio ethylbenzene/CH3CN of 1/1. These results are objects of two patent applications [60,61].
Light-induced activation
The first example of light-induced in situ generation of PINO radical was reported in 2007 by Lucarini and co-workers [62]. Irradiation of N-alkoxyphthalimides with filtered light (λ > 300 nm) from a mercury lamp promoted the selective homolysis of the O–C bond, leading to the formation of the N-oxyl radical, as documented by the strong characteristic EPR signal. The efficiency of the initiation approach was documented by measuring the dioxygen consumption during the aerobic oxidation of cumene. In the absence of light, cumene was completely inert, thus proving the intervention of the catalysts in the oxidative cycle.
This process was successfully employed in the selective oxidation of benzyl alcohol at room temperature, affording the corresponding aldehyde in 4 h, in accordance with the results previously reported by our group operating under NHPI/Co(OAc)2 catalysis [63,64] (Scheme 22).
![[1860-5397-9-146-i22]](/bjoc/content/inline/1860-5397-9-146-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Light-induced generation of PINO from N-alkoxyphthalimides.
Scheme 22: Light-induced generation of PINO from N-alkoxyphthalimides.
Even if this procedure seems to be particularly valuable, as no further radical mediators or initiators are required, it cannot be applied directly to NHPI.
More recently, Antonietti and co-workers have reported the photocatalytic oxidative activation of NHPI by graphitic carbon nitride (g-C3N4) and visible light irradiation [65].
g-C3N4, the most stable allotrope of carbon nitride, is a two-dimensional polymer with a tri-s-triazine ring unit and a π-conjugated layered structure similar to graphene. It is a medium-band-gap semiconductor and has proved to be an efficient photocatalyst for synthetic purposes [66,67].
It has been demonstrated that the excited state of g-C3N4, obtained by irradiation with visible light, is able to activate O2 to the corresponding superoxide radical. The latter could undergo hydrogen abstraction from NHPI, leading to the generation of PINO.
This approach was successfully applied to promote the visible-light-induced metal-free oxidation of allylic substrates (Scheme 23).
![[1860-5397-9-146-i23]](/bjoc/content/inline/1860-5397-9-146-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Visible-light/g-C3N4 induced metal-free oxidation of allylic substrates.
Scheme 23: Visible-light/g-C3N4 induced metal-free oxidation of allylic substrates.
Other initiators
In their ongoing research dedicated to NHPI activation towards the aerobic oxidation of hydrocarbons, Xu and co-workers developed other redox initiators in addition to the NHPI/quinone systems previously described.
In 2005 they reported the selective oxygenation of ethylbenzene to the corresponding acetophenone by means of a NHPI/o-phenanthroline-mediated organocatalytic system, in the presence of molecular bromine as a co-catalyst [68] (Scheme 24).
![[1860-5397-9-146-i24]](/bjoc/content/inline/1860-5397-9-146-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: NHPI/o-phenanthroline-mediated organocatalytic system.
Scheme 24: NHPI/o-phenanthroline-mediated organocatalytic system.
According to the proposed mechanism, Br2 generates ET processes by oxidizing the o-phenanthroline to the corresponding cation radicals. The latter promote in turn ET and HAT processes with NHPI, leading to the formation of PINO.
In 2009 the same research group also performed the oxidation of ethylbenzene and other hydrocarbons by combining NHPI with alkaline-earth chlorides under an aerobic atmosphere [69]. The intervention of the PINO radical in the oxidation mechanism was confirmed by UV–vis and FTIR spectroscopy. MgCl2 was found to be the most effective salt among those tested for this purpose.
An efficient and selective aerobic oxidation of hydrocarbons to their oxygenated products was also achieved by Zheng et al. by combining NHPI with dimethylglyoxime (DMG) [70]. The suggested mechanism is depicted in Scheme 25.
![[1860-5397-9-146-i25]](/bjoc/content/inline/1860-5397-9-146-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: NHPI/DMG-mediated organocatalytic system.
Scheme 25: NHPI/DMG-mediated organocatalytic system.
Once again all the approaches described above led to the formation of acetophenone as the major product of ethylbenzene oxidation.
A different result was achieved by Fierro et al. who, in the same year, reported the ethylbenzene oxidation to its hydroperoxide by operating in the presence of NHPI (or other NHIs, i.e., N-hydroxysuccinimide, N-hydroxymaleimide and N-hydroxynaphthalimide) and tiny amounts of sodium hydroxide (5 × 10−3 mol % with respect to ethylbenzene) at 421 K and 0.3 MPa [71]. The addition of NaOH determined a significant increase of selectivity in hydroperoxide (77.3%), while the ethylbenzene conversion slightly decreased from 24.5 to 21.3%.
Very recently NHPI was reported to catalyze selective transformations without requiring additional initiators. Jiao et al. described the catalyzed oxidative cleavage of C=C double bonds using molecular oxygen as the final oxidant [72]. Following this approach α-methylstyrene was converted to acetophenone in 80% yield by operating at 80 °C in N,N-dimethylacetamide (DMA) as solvent. A plausible mechanism is shown in Scheme 26. It is based on three experimental data sets collected by the research group: (i) The occurrence of a free-radical initiation. In fact, in the presence of the radical scavenger BHT no conversion was observed. (ii) The exclusion that epoxide could be an intermediate of the process. When 2-methyl-2-phenyloxirane was used in place of α-methylstyrene, poor results were obtained in terms of yield in acetophenone. (iii) The proof that the oxygen atom in acetophenone originated from molecular oxygen, by conducting the oxygenation in the presence of 18O2.
![[1860-5397-9-146-i26]](/bjoc/content/inline/1860-5397-9-146-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: NHPI catalyzed oxidative cleavage of C=C bonds.
Scheme 26: NHPI catalyzed oxidative cleavage of C=C bonds.
Finally, Inoue and co-workers reported an efficient C(sp3)–N bond-forming method consisting of the chemoselective conversion of C(sp3)–H bonds in the presence of stoichiometric amounts of dialkyl azodicarboxylate reagent [73]. The proposed mechanism (Scheme 27) suggests a double function of the dialkyl azodicarboxylate, which acts both as an oxidant, promoting the oxidation of NHPI to PINO by ET process, and as acceptor of carbon radicals generated via HAT by PINO, leading to the formation of hydrazine derivatives. The scope of the reaction was investigated and its applicability was extended to different C–H bonds, including benzylic, propargylic, and aliphatic ones.
![[1860-5397-9-146-i27]](/bjoc/content/inline/1860-5397-9-146-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of hydrazine derivatives.
Scheme 27: Synthesis of hydrazine derivatives.
Furthermore, the hydrazine compounds were readily converted to the corresponding carbamates and amines.
Conclusion
Metal-free oxidation catalyzed by N-hydroxyphthalimide and analogous N-hydroxy imides is becoming an essential tool for all research groups directly involved in the development of selective transformations mediated by molecular oxygen. This increasing interest is testified by several papers that have appeared in the past few years in this field and are herein discussed, describing intriguing free-radical routes for the generation of N-oxyl active species under mild conditions. The fields of application range from large-scale production, such as the selective oxidation of alkyl aromatics, to the synthesis of fine chemicals.
Metal-free PINO activation represents another decisive step towards the consecration of NHPI as the mediator of first choice to perform green and selective aerobic oxidations. The next one would be the engineering of valuable solutions for the complete recovery and recycle of this homogeneous organocatalyst. Once reached, this final goal, combined with the use of molecular oxygen as stoichiometric oxidant under mild operative conditions, would definitely open the way towards much more cost-effective and environmentally friendly oxidative processes.
References
-
Bäckvall, J.-E., Ed. Modern Oxidation Methods; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. doi:10.1002/3527603689
Return to citation in text: [1] -
Hermans, I.; Peeters, J.; Jacobs, P. A. Top. Catal. 2008, 48, 41–48. doi:10.1007/s11244-008-9051-x
Return to citation in text: [1] -
Hermans, I.; Peeters, J.; Jacobs, P. A. Top. Catal. 2008, 50, 124–132. doi:10.1007/s11244-008-9099-7
Return to citation in text: [1] -
Hermans, I.; Spier, E. S.; Neuenschwander, U.; Turrà, N.; Baiker, A. Top. Catal. 2009, 52, 1162–1174. doi:10.1007/s11244-009-9268-3
Return to citation in text: [1] -
Gambarotti, C.; Melone, L.; Caronna, T.; Punta, C. Curr. Org. Chem., in press.
Return to citation in text: [1] -
Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Chem. Rev. 2005, 105, 2329–2364. doi:10.1021/cr050523v
Return to citation in text: [1] -
Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k
Return to citation in text: [1] -
Galli, C.; Gentili, P.; Lanzalunga, O. Angew. Chem., Int. Ed. 2008, 47, 4790–4796. doi:10.1002/anie.200704292
Return to citation in text: [1] -
Coseri, S. Mini-Rev. Org. Chem. 2008, 5, 222–227. doi:10.2174/157019308785161675
Return to citation in text: [1] -
Coseri, S. Catal. Rev. - Sci. Eng. 2009, 51, 218–292. doi:10.1080/01614940902743841
Return to citation in text: [1] -
Punta, C.; Gambarotti, C. N-Hydroxy Derivatives: Key Organocatalysts for the Selective Free Radical Aerobic Oxidation of Organic Compounds. In Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry; Pignataro, B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp 3–24. doi:10.1002/9783527630554.ch1
Return to citation in text: [1] -
Amorati, R.; Lucarini, M.; Mugnaini, M.; Pedulli, G. F.; Minisci, F.; Recupero, F.; Fontana, F.; Astolfi, P.; Greci, L. J. Org. Chem. 2003, 68, 1747–1754. doi:10.1021/jo026660z
Return to citation in text: [1] [2] [3] -
Minisci, F.; Recupero, F.; Pedulli, G. F.; Lucarini, M. J. Mol. Catal. A 2003, 204–205, 63–90. doi:10.1016/S1381-1169(03)00286-3
Return to citation in text: [1] -
Tsujimoto, S.; Iwahama, T.; Sakaguchi, S.; Ishii, Y. Chem. Commun. 2001, 2352–2353. doi:10.1039/b107548a
Return to citation in text: [1] -
Tsujimoto, S.; Sakaguchi, S.; Ishii, Y. Tetrahedron Lett. 2003, 44, 5601–5604. doi:10.1016/S0040-4039(03)01375-3
Return to citation in text: [1] [2] -
Aoki, Y.; Sakaguchi, S.; Ishii, Y. Adv. Synth. Catal. 2004, 346, 199–202. doi:10.1002/adsc.200303168
Return to citation in text: [1] -
Aoki, Y.; Hirai, N.; Sakaguchi, S.; Ishii, Y. Tetrahedron 2005, 61, 10995–10999. doi:10.1016/j.tet.2005.08.087
Return to citation in text: [1] -
Arends, I. W. C. E.; Sasidharan, M.; Kühnle, A.; Duda, M.; Jost, C.; Sheldon, R. A. Tetrahedron 2002, 58, 9055–9061. doi:10.1016/S0040-4020(02)01131-6
Return to citation in text: [1] -
Punta, C.; Rector, C. L.; Porter, N. A. Chem. Res. Toxicol. 2005, 18, 349–356. doi:10.1021/tx049685x
Return to citation in text: [1] [2] -
Porter, N. A. J. Org. Chem. 2013, 78, 3511–3524. doi:10.1021/jo4001433
Return to citation in text: [1] -
Schmidt, V. A.; Alexanian, E. J. Angew. Chem., Int. Ed. 2010, 49, 4491–4494. doi:10.1002/anie.201000843
Return to citation in text: [1] -
Sakaguchi, S.; Eikawa, M.; Ishii, Y. Tetrahedron Lett. 1997, 38, 7075–7078. doi:10.1016/S0040-4039(97)01652-3
Return to citation in text: [1] -
Eikawa, M.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 1999, 64, 4676–4679. doi:10.1021/jo982406o
Return to citation in text: [1] -
Sakaguchi, S.; Nishiwaki, Y.; Kitamura, T.; Ishii, Y. Angew. Chem., Int. Ed. 2001, 40, 222–224. doi:10.1002/1521-3773(20010105)40:1<222::AID-ANIE222>3.0.CO;2-W
Return to citation in text: [1] -
Nishiwaki, Y.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2002, 67, 5663–5668. doi:10.1021/jo025632d
Return to citation in text: [1] -
Minisci, F.; Recupero, F.; Gambarotti, C.; Punta, C.; Paganelli, R. Tetrahedron Lett. 2003, 44, 6919–6922. doi:10.1016/S0040-4039(03)01687-3
Return to citation in text: [1] -
Fabbrini, M.; Galli, C.; Gentili, P. J. Mol. Catal. B: Enzym. 2002, 16, 231–240. doi:10.1016/S1381-1177(01)00067-4
Return to citation in text: [1] [2] -
Baiocco, P.; Barreca, A. M.; Fabbrini, M.; Galli, C.; Gentili, P. Org. Biomol. Chem. 2003, 1, 191–197. doi:10.1039/b208951c
Return to citation in text: [1] [2] -
Branchi, B.; Galli, C.; Gentili, P. Org. Biomol. Chem. 2005, 3, 2604–2614. doi:10.1039/b504199f
Return to citation in text: [1] -
Cantarella, G.; Galli, C.; Gentili, P. New J. Chem. 2004, 28, 366–372. doi:10.1039/b311907f
Return to citation in text: [1] -
Astolfi, P.; Brandi, P.; Galli, C.; Gentili, P.; Gerini, M. F.; Greci, L.; Lanzalunga, O. New J. Chem. 2005, 29, 1308–1317. doi:10.1039/b507657a
Return to citation in text: [1] -
d'Acunzo, F.; Baiocco, P.; Galli, C. New J. Chem. 2003, 27, 329–332. doi:10.1039/b210978f
Return to citation in text: [1] -
Yang, G.; Ma, Y.; Xu, J. J. Am. Chem. Soc. 2004, 126, 10542–10543. doi:10.1021/ja047297b
Return to citation in text: [1] [2] -
Yang, G.; Zhang, Q.; Miao, H.; Tong, X.; Xu, J. Org. Lett. 2005, 7, 263–266. doi:10.1021/ol047749p
Return to citation in text: [1] -
Zhou, L.; Chen, Y.; Yang, X.; Su, Y.; Zhang, W.; Xu, J. Catal. Lett. 2008, 125, 154–159. doi:10.1007/s10562-008-9537-y
Return to citation in text: [1] -
Zhang, Q.; Chen, C.; Ma, H.; Miao, H.; Zhang, W.; Sun, Z.; Xu, J. J. Chem. Technol. Biotechnol. 2008, 83, 1364–1369. doi:10.1002/jctb.1977
Return to citation in text: [1] -
Yang, X.; Wang, Y.; Zhou, L.; Chen, C.; Zhang, W.; Xu, J. J. Chem. Technol. Biotechnol. 2010, 85, 564–568. doi:10.1002/jctb.2345
Return to citation in text: [1] -
Du, Z.; Sun, Z.; Zhang, W.; Miao, H.; Ma, H.; Xu, J. Tetrahedron Lett. 2009, 50, 1677–1680. doi:10.1016/j.tetlet.2009.01.077
Return to citation in text: [1] -
Hermans, I.; Vereecken, L.; Jacobs, P. A.; Peeters, J. Chem. Commun. 2004, 1140–1141. doi:10.1039/b401050g
Return to citation in text: [1] -
Hermans, I.; Jacobs, P. A.; Peeters, J. Phys. Chem. Chem. Phys. 2007, 9, 686–690. doi:10.1039/b616392k
Return to citation in text: [1] -
Hermans, I.; Jacobs, P. A.; Peeters, J. Phys. Chem. Chem. Phys. 2008, 10, 1125–1132. doi:10.1039/b716932a
Return to citation in text: [1] -
Wang, C.; Wang, G.; Mao, J.; Yao, Z.; Li, H. Catal. Commun. 2010, 11, 758–762. doi:10.1016/j.catcom.2010.02.010
Return to citation in text: [1] -
Chen, K.; Sun, Y.; Wang, C.; Yao, J.; Chen, Z.; Li, H. Phys. Chem. Chem. Phys. 2012, 14, 12141–12146. doi:10.1039/c2cp41617d
Return to citation in text: [1] -
Coseri, S.; Nistor, G.; Fras, L.; Strnad, S.; Harabagiu, V.; Simionescu, B. C. Biomacromolecules 2009, 10, 2294–2299. doi:10.1021/bm9004854
Return to citation in text: [1] -
Biliuta, G.; Fras, L.; Strnad, S.; Harabagiu, V.; Coseri, S. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 4790–4799. doi:10.1002/pola.24270
Return to citation in text: [1] -
Coseri, S.; Biliuta, G.; Simionescu, B. C.; Stana-Kleinschek, K.; Ribitsch, V.; Harabagiu, V. Carbohydr. Polym. 2013, 93, 207–215. doi:10.1016/j.carbpol.2012.03.086
Return to citation in text: [1] -
Melone, L.; Altomare, L.; Alfieri, I.; Lorenzi, A.; De Nardo, L.; Punta, C. J. Photochem. Photobiol., A 2013, 261, 53–60. doi:10.1016/j.jphotochem.2013.04.004.
Return to citation in text: [1] -
Biliuta, G.; Fras, L.; Drobota, M.; Persin, Z.; Kreze, T.; Stana-Kleinschek, K.; Ribitsch, V.; Harabagiu, V.; Coseri, S. Carbohydr. Polym. 2013, 91, 502–507. doi:10.1016/j.carbpol.2012.08.047
Return to citation in text: [1] -
Biliuta, G.; Fras, L.; Harabagiu, V.; Coseri, S. Digest J. Nanomat. Biostruct. 2011, 6, 291–297.
Return to citation in text: [1] -
Melone, L.; Punta, C. Co-oxidation processes promoted by N-hydroxyphthalimide/aldehyde system. In New Developments in Aldehydes Research; Torrioni, L.; Pescasseroni, E., Eds.; Nova Publisher, 2013; pp 121–138.
Return to citation in text: [1] -
Einhorn, C.; Einhorn, J.; Marcadal, C.; Pierre, J.-L. Chem. Commun. 1997, 447–448. doi:10.1039/a607463d
Return to citation in text: [1] -
Minisci, F.; Gambarotti, C.; Pierini, M.; Porta, O.; Punta, C.; Recupero, F.; Lucarini, M.; Mugnaini, V. Tetrahedron Lett. 2006, 47, 1421–1424. doi:10.1016/j.tetlet.2005.12.089
Return to citation in text: [1] [2] [3] -
Bravo, A.; Bjørsvik, H.-R.; Fontana, F.; Minisci, F.; Serri, A. J. Org. Chem. 1996, 61, 9409–9416. doi:10.1021/jo961366q
Return to citation in text: [1] -
Hermans, I.; Jacobs, P. A.; Peeters, J. Chem.–Eur. J. 2006, 12, 4229–4240. doi:10.1002/chem.200600189
Return to citation in text: [1] -
Turrà, N.; Neuenschwander, U.; Hermans, I. ChemPhysChem 2013, 14, 1666–1669. doi:10.1002/cphc.201300130.
Return to citation in text: [1] -
Punta, C.; Moscatelli, D.; Porta, O.; Minisci, F.; Gambarotti, C.; Lucarini, M. Selective aerobic radical epoxidation of α-olefins catalyzed by N-Hydroxyphthalimide. In Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis; Oyama, S. T., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp 217–229. doi:10.1016/B978-0-444-53188-9.00006-7
Return to citation in text: [1] -
Spaccini, R.; Liguori, L.; Punta, C.; Bjørsvik, H.-R. ChemSusChem 2012, 5, 261–265. doi:10.1002/cssc.201100262
Return to citation in text: [1] -
Melone, L.; Gambarotti, C.; Prosperini, S.; Pastori, N.; Recupero, F.; Punta, C. Adv. Synth. Catal. 2011, 353, 147–154. doi:10.1002/adsc.201000786
Return to citation in text: [1] -
Melone, L.; Prosperini, S.; Gambarotti, C.; Pastori, N.; Recupero, F.; Punta, C. J. Mol. Catal. A 2012, 355, 155–160. doi:10.1016/j.molcata.2011.12.009
Return to citation in text: [1] -
Minisci, F.; Porta, O.; Recupero, F.; Punta, C.; Gambarotti, C.; Pierini, M. Process for the preparation of phenol by means of new catalytic systems. WO2008037435, April 3, 2008.
Return to citation in text: [1] -
Minisci, F.; Porta, O.; Recupero, F.; Punta, C.; Gambarotti, C.; Spaccini, R. Catalytic process for the preparation of hydroperoxides of alkylbenzenes by aerobic oxidation under mild conditions. WO2009115275, Sept 24, 2009.
Return to citation in text: [1] -
Lucarini, M.; Ferroni, F.; Pedulli, G. F.; Gardi, S.; Lazzari, D.; Schlingloff, G.; Sala, M. Tetrahedron Lett. 2007, 48, 5331–5334. doi:10.1016/j.tetlet.2007.05.162
Return to citation in text: [1] -
Minisci, F.; Punta, C.; Recupero, F.; Fontana, F.; Pedulli, G. F. Chem. Commun. 2002, 688–689. doi:10.1039/b110451a
Return to citation in text: [1] -
Minisci, F.; Recupero, F.; Cecchetto, A.; Gambarotti, C.; Punta, C.; Faletti, R.; Paganelli, R.; Pedulli, G. F. Eur. J. Org. Chem. 2004, 109–119. doi:10.1002/ejoc.200300332
Return to citation in text: [1] -
Zhang, P.; Wang, Y.; Yao, J.; Wang, C.; Yan, C.; Antonietti, M.; Li, H. Adv. Synth. Catal. 2011, 353, 1447–1451. doi:10.1002/adsc.201100175
Return to citation in text: [1] -
Wang, Y.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2012, 51, 68–89. doi:10.1002/anie.201101182
Return to citation in text: [1] -
Wang, X.; Blechert, S.; Antonietti, M. ACS Catal. 2012, 2, 1596–1606. doi:10.1021/cs300240x
Return to citation in text: [1] -
Tong, X.; Xu, J.; Miao, H. Adv. Synth. Catal. 2005, 347, 1953–1957. doi:10.1002/adsc.200505183
Return to citation in text: [1] -
Yang, X.; Zhou, L.; Chen, Y.; Chen, C.; Su, Y.; Miao, H.; Xu, J. Catal. Commun. 2009, 11, 171–174. doi:10.1016/j.catcom.2009.09.019
Return to citation in text: [1] -
Zheng, G.; Liu, C.; Wang, Q.; Wang, M.; Yang, G. Adv. Synth. Catal. 2009, 351, 2638–2642. doi:10.1002/adsc.200900509
Return to citation in text: [1] -
Toribio, P. P.; Gimeno-Gargallo, A.; Capel-Sanchez, M. C.; de Frutos, M. P.; Campos-Martin, J. M.; Fierro, J. L. G. Appl. Catal., A 2009, 363, 32–39. doi:10.1016/j.apcata.2009.04.023
Return to citation in text: [1] -
Lin, R.; Chen, F.; Jiao, N. Org. Lett. 2012, 14, 4158–4161. doi:10.1021/ol3018215
Return to citation in text: [1] -
Amaoka, Y.; Kamijio, S.; Hoshikawa, T.; Inoue, M. J. Org. Chem. 2012, 77, 9959–9969. doi:10.1021/jo301840e
Return to citation in text: [1]
| 33. | Yang, G.; Ma, Y.; Xu, J. J. Am. Chem. Soc. 2004, 126, 10542–10543. doi:10.1021/ja047297b |
| 34. | Yang, G.; Zhang, Q.; Miao, H.; Tong, X.; Xu, J. Org. Lett. 2005, 7, 263–266. doi:10.1021/ol047749p |
| 35. | Zhou, L.; Chen, Y.; Yang, X.; Su, Y.; Zhang, W.; Xu, J. Catal. Lett. 2008, 125, 154–159. doi:10.1007/s10562-008-9537-y |
| 45. | Biliuta, G.; Fras, L.; Strnad, S.; Harabagiu, V.; Coseri, S. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 4790–4799. doi:10.1002/pola.24270 |
| 46. | Coseri, S.; Biliuta, G.; Simionescu, B. C.; Stana-Kleinschek, K.; Ribitsch, V.; Harabagiu, V. Carbohydr. Polym. 2013, 93, 207–215. doi:10.1016/j.carbpol.2012.03.086 |
| 47. | Melone, L.; Altomare, L.; Alfieri, I.; Lorenzi, A.; De Nardo, L.; Punta, C. J. Photochem. Photobiol., A 2013, 261, 53–60. doi:10.1016/j.jphotochem.2013.04.004. |
| 42. | Wang, C.; Wang, G.; Mao, J.; Yao, Z.; Li, H. Catal. Commun. 2010, 11, 758–762. doi:10.1016/j.catcom.2010.02.010 |
| 43. | Chen, K.; Sun, Y.; Wang, C.; Yao, J.; Chen, Z.; Li, H. Phys. Chem. Chem. Phys. 2012, 14, 12141–12146. doi:10.1039/c2cp41617d |
| 44. | Coseri, S.; Nistor, G.; Fras, L.; Strnad, S.; Harabagiu, V.; Simionescu, B. C. Biomacromolecules 2009, 10, 2294–2299. doi:10.1021/bm9004854 |
| 38. | Du, Z.; Sun, Z.; Zhang, W.; Miao, H.; Ma, H.; Xu, J. Tetrahedron Lett. 2009, 50, 1677–1680. doi:10.1016/j.tetlet.2009.01.077 |
| 39. | Hermans, I.; Vereecken, L.; Jacobs, P. A.; Peeters, J. Chem. Commun. 2004, 1140–1141. doi:10.1039/b401050g |
| 40. | Hermans, I.; Jacobs, P. A.; Peeters, J. Phys. Chem. Chem. Phys. 2007, 9, 686–690. doi:10.1039/b616392k |
| 41. | Hermans, I.; Jacobs, P. A.; Peeters, J. Phys. Chem. Chem. Phys. 2008, 10, 1125–1132. doi:10.1039/b716932a |
| 36. | Zhang, Q.; Chen, C.; Ma, H.; Miao, H.; Zhang, W.; Sun, Z.; Xu, J. J. Chem. Technol. Biotechnol. 2008, 83, 1364–1369. doi:10.1002/jctb.1977 |
| 37. | Yang, X.; Wang, Y.; Zhou, L.; Chen, C.; Zhang, W.; Xu, J. J. Chem. Technol. Biotechnol. 2010, 85, 564–568. doi:10.1002/jctb.2345 |
| 48. | Biliuta, G.; Fras, L.; Drobota, M.; Persin, Z.; Kreze, T.; Stana-Kleinschek, K.; Ribitsch, V.; Harabagiu, V.; Coseri, S. Carbohydr. Polym. 2013, 91, 502–507. doi:10.1016/j.carbpol.2012.08.047 |
| 49. | Biliuta, G.; Fras, L.; Harabagiu, V.; Coseri, S. Digest J. Nanomat. Biostruct. 2011, 6, 291–297. |
| 50. | Melone, L.; Punta, C. Co-oxidation processes promoted by N-hydroxyphthalimide/aldehyde system. In New Developments in Aldehydes Research; Torrioni, L.; Pescasseroni, E., Eds.; Nova Publisher, 2013; pp 121–138. |
| 57. | Spaccini, R.; Liguori, L.; Punta, C.; Bjørsvik, H.-R. ChemSusChem 2012, 5, 261–265. doi:10.1002/cssc.201100262 |
| 58. | Melone, L.; Gambarotti, C.; Prosperini, S.; Pastori, N.; Recupero, F.; Punta, C. Adv. Synth. Catal. 2011, 353, 147–154. doi:10.1002/adsc.201000786 |
| 59. | Melone, L.; Prosperini, S.; Gambarotti, C.; Pastori, N.; Recupero, F.; Punta, C. J. Mol. Catal. A 2012, 355, 155–160. doi:10.1016/j.molcata.2011.12.009 |
| 52. | Minisci, F.; Gambarotti, C.; Pierini, M.; Porta, O.; Punta, C.; Recupero, F.; Lucarini, M.; Mugnaini, V. Tetrahedron Lett. 2006, 47, 1421–1424. doi:10.1016/j.tetlet.2005.12.089 |
| 56. | Punta, C.; Moscatelli, D.; Porta, O.; Minisci, F.; Gambarotti, C.; Lucarini, M. Selective aerobic radical epoxidation of α-olefins catalyzed by N-Hydroxyphthalimide. In Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis; Oyama, S. T., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp 217–229. doi:10.1016/B978-0-444-53188-9.00006-7 |
| 53. | Bravo, A.; Bjørsvik, H.-R.; Fontana, F.; Minisci, F.; Serri, A. J. Org. Chem. 1996, 61, 9409–9416. doi:10.1021/jo961366q |
| 54. | Hermans, I.; Jacobs, P. A.; Peeters, J. Chem.–Eur. J. 2006, 12, 4229–4240. doi:10.1002/chem.200600189 |
| 55. | Turrà, N.; Neuenschwander, U.; Hermans, I. ChemPhysChem 2013, 14, 1666–1669. doi:10.1002/cphc.201300130. |
| 52. | Minisci, F.; Gambarotti, C.; Pierini, M.; Porta, O.; Punta, C.; Recupero, F.; Lucarini, M.; Mugnaini, V. Tetrahedron Lett. 2006, 47, 1421–1424. doi:10.1016/j.tetlet.2005.12.089 |
| 51. | Einhorn, C.; Einhorn, J.; Marcadal, C.; Pierre, J.-L. Chem. Commun. 1997, 447–448. doi:10.1039/a607463d |
| 52. | Minisci, F.; Gambarotti, C.; Pierini, M.; Porta, O.; Punta, C.; Recupero, F.; Lucarini, M.; Mugnaini, V. Tetrahedron Lett. 2006, 47, 1421–1424. doi:10.1016/j.tetlet.2005.12.089 |
| 62. | Lucarini, M.; Ferroni, F.; Pedulli, G. F.; Gardi, S.; Lazzari, D.; Schlingloff, G.; Sala, M. Tetrahedron Lett. 2007, 48, 5331–5334. doi:10.1016/j.tetlet.2007.05.162 |
| 63. | Minisci, F.; Punta, C.; Recupero, F.; Fontana, F.; Pedulli, G. F. Chem. Commun. 2002, 688–689. doi:10.1039/b110451a |
| 64. | Minisci, F.; Recupero, F.; Cecchetto, A.; Gambarotti, C.; Punta, C.; Faletti, R.; Paganelli, R.; Pedulli, G. F. Eur. J. Org. Chem. 2004, 109–119. doi:10.1002/ejoc.200300332 |
| 60. | Minisci, F.; Porta, O.; Recupero, F.; Punta, C.; Gambarotti, C.; Pierini, M. Process for the preparation of phenol by means of new catalytic systems. WO2008037435, April 3, 2008. |
| 61. | Minisci, F.; Porta, O.; Recupero, F.; Punta, C.; Gambarotti, C.; Spaccini, R. Catalytic process for the preparation of hydroperoxides of alkylbenzenes by aerobic oxidation under mild conditions. WO2009115275, Sept 24, 2009. |
| 1. | Bäckvall, J.-E., Ed. Modern Oxidation Methods; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. doi:10.1002/3527603689 |
| 12. | Amorati, R.; Lucarini, M.; Mugnaini, M.; Pedulli, G. F.; Minisci, F.; Recupero, F.; Fontana, F.; Astolfi, P.; Greci, L. J. Org. Chem. 2003, 68, 1747–1754. doi:10.1021/jo026660z |
| 12. | Amorati, R.; Lucarini, M.; Mugnaini, M.; Pedulli, G. F.; Minisci, F.; Recupero, F.; Fontana, F.; Astolfi, P.; Greci, L. J. Org. Chem. 2003, 68, 1747–1754. doi:10.1021/jo026660z |
| 72. | Lin, R.; Chen, F.; Jiao, N. Org. Lett. 2012, 14, 4158–4161. doi:10.1021/ol3018215 |
| 7. | Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k |
| 8. | Galli, C.; Gentili, P.; Lanzalunga, O. Angew. Chem., Int. Ed. 2008, 47, 4790–4796. doi:10.1002/anie.200704292 |
| 9. | Coseri, S. Mini-Rev. Org. Chem. 2008, 5, 222–227. doi:10.2174/157019308785161675 |
| 10. | Coseri, S. Catal. Rev. - Sci. Eng. 2009, 51, 218–292. doi:10.1080/01614940902743841 |
| 11. | Punta, C.; Gambarotti, C. N-Hydroxy Derivatives: Key Organocatalysts for the Selective Free Radical Aerobic Oxidation of Organic Compounds. In Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry; Pignataro, B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp 3–24. doi:10.1002/9783527630554.ch1 |
| 19. | Punta, C.; Rector, C. L.; Porter, N. A. Chem. Res. Toxicol. 2005, 18, 349–356. doi:10.1021/tx049685x |
| 6. | Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Chem. Rev. 2005, 105, 2329–2364. doi:10.1021/cr050523v |
| 19. | Punta, C.; Rector, C. L.; Porter, N. A. Chem. Res. Toxicol. 2005, 18, 349–356. doi:10.1021/tx049685x |
| 70. | Zheng, G.; Liu, C.; Wang, Q.; Wang, M.; Yang, G. Adv. Synth. Catal. 2009, 351, 2638–2642. doi:10.1002/adsc.200900509 |
| 2. | Hermans, I.; Peeters, J.; Jacobs, P. A. Top. Catal. 2008, 48, 41–48. doi:10.1007/s11244-008-9051-x |
| 3. | Hermans, I.; Peeters, J.; Jacobs, P. A. Top. Catal. 2008, 50, 124–132. doi:10.1007/s11244-008-9099-7 |
| 4. | Hermans, I.; Spier, E. S.; Neuenschwander, U.; Turrà, N.; Baiker, A. Top. Catal. 2009, 52, 1162–1174. doi:10.1007/s11244-009-9268-3 |
| 5. | Gambarotti, C.; Melone, L.; Caronna, T.; Punta, C. Curr. Org. Chem., in press. |
| 71. | Toribio, P. P.; Gimeno-Gargallo, A.; Capel-Sanchez, M. C.; de Frutos, M. P.; Campos-Martin, J. M.; Fierro, J. L. G. Appl. Catal., A 2009, 363, 32–39. doi:10.1016/j.apcata.2009.04.023 |
| 15. | Tsujimoto, S.; Sakaguchi, S.; Ishii, Y. Tetrahedron Lett. 2003, 44, 5601–5604. doi:10.1016/S0040-4039(03)01375-3 |
| 17. | Aoki, Y.; Hirai, N.; Sakaguchi, S.; Ishii, Y. Tetrahedron 2005, 61, 10995–10999. doi:10.1016/j.tet.2005.08.087 |
| 68. | Tong, X.; Xu, J.; Miao, H. Adv. Synth. Catal. 2005, 347, 1953–1957. doi:10.1002/adsc.200505183 |
| 14. | Tsujimoto, S.; Iwahama, T.; Sakaguchi, S.; Ishii, Y. Chem. Commun. 2001, 2352–2353. doi:10.1039/b107548a |
| 15. | Tsujimoto, S.; Sakaguchi, S.; Ishii, Y. Tetrahedron Lett. 2003, 44, 5601–5604. doi:10.1016/S0040-4039(03)01375-3 |
| 18. | Arends, I. W. C. E.; Sasidharan, M.; Kühnle, A.; Duda, M.; Jost, C.; Sheldon, R. A. Tetrahedron 2002, 58, 9055–9061. doi:10.1016/S0040-4020(02)01131-6 |
| 69. | Yang, X.; Zhou, L.; Chen, Y.; Chen, C.; Su, Y.; Miao, H.; Xu, J. Catal. Commun. 2009, 11, 171–174. doi:10.1016/j.catcom.2009.09.019 |
| 12. | Amorati, R.; Lucarini, M.; Mugnaini, M.; Pedulli, G. F.; Minisci, F.; Recupero, F.; Fontana, F.; Astolfi, P.; Greci, L. J. Org. Chem. 2003, 68, 1747–1754. doi:10.1021/jo026660z |
| 65. | Zhang, P.; Wang, Y.; Yao, J.; Wang, C.; Yan, C.; Antonietti, M.; Li, H. Adv. Synth. Catal. 2011, 353, 1447–1451. doi:10.1002/adsc.201100175 |
| 13. | Minisci, F.; Recupero, F.; Pedulli, G. F.; Lucarini, M. J. Mol. Catal. A 2003, 204–205, 63–90. doi:10.1016/S1381-1169(03)00286-3 |
| 16. | Aoki, Y.; Sakaguchi, S.; Ishii, Y. Adv. Synth. Catal. 2004, 346, 199–202. doi:10.1002/adsc.200303168 |
| 66. | Wang, Y.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2012, 51, 68–89. doi:10.1002/anie.201101182 |
| 67. | Wang, X.; Blechert, S.; Antonietti, M. ACS Catal. 2012, 2, 1596–1606. doi:10.1021/cs300240x |
| 23. | Eikawa, M.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 1999, 64, 4676–4679. doi:10.1021/jo982406o |
| 21. | Schmidt, V. A.; Alexanian, E. J. Angew. Chem., Int. Ed. 2010, 49, 4491–4494. doi:10.1002/anie.201000843 |
| 22. | Sakaguchi, S.; Eikawa, M.; Ishii, Y. Tetrahedron Lett. 1997, 38, 7075–7078. doi:10.1016/S0040-4039(97)01652-3 |
| 73. | Amaoka, Y.; Kamijio, S.; Hoshikawa, T.; Inoue, M. J. Org. Chem. 2012, 77, 9959–9969. doi:10.1021/jo301840e |
| 32. | d'Acunzo, F.; Baiocco, P.; Galli, C. New J. Chem. 2003, 27, 329–332. doi:10.1039/b210978f |
| 33. | Yang, G.; Ma, Y.; Xu, J. J. Am. Chem. Soc. 2004, 126, 10542–10543. doi:10.1021/ja047297b |
| 29. | Branchi, B.; Galli, C.; Gentili, P. Org. Biomol. Chem. 2005, 3, 2604–2614. doi:10.1039/b504199f |
| 27. | Fabbrini, M.; Galli, C.; Gentili, P. J. Mol. Catal. B: Enzym. 2002, 16, 231–240. doi:10.1016/S1381-1177(01)00067-4 |
| 28. | Baiocco, P.; Barreca, A. M.; Fabbrini, M.; Galli, C.; Gentili, P. Org. Biomol. Chem. 2003, 1, 191–197. doi:10.1039/b208951c |
| 30. | Cantarella, G.; Galli, C.; Gentili, P. New J. Chem. 2004, 28, 366–372. doi:10.1039/b311907f |
| 31. | Astolfi, P.; Brandi, P.; Galli, C.; Gentili, P.; Gerini, M. F.; Greci, L.; Lanzalunga, O. New J. Chem. 2005, 29, 1308–1317. doi:10.1039/b507657a |
| 26. | Minisci, F.; Recupero, F.; Gambarotti, C.; Punta, C.; Paganelli, R. Tetrahedron Lett. 2003, 44, 6919–6922. doi:10.1016/S0040-4039(03)01687-3 |
| 27. | Fabbrini, M.; Galli, C.; Gentili, P. J. Mol. Catal. B: Enzym. 2002, 16, 231–240. doi:10.1016/S1381-1177(01)00067-4 |
| 28. | Baiocco, P.; Barreca, A. M.; Fabbrini, M.; Galli, C.; Gentili, P. Org. Biomol. Chem. 2003, 1, 191–197. doi:10.1039/b208951c |
| 24. | Sakaguchi, S.; Nishiwaki, Y.; Kitamura, T.; Ishii, Y. Angew. Chem., Int. Ed. 2001, 40, 222–224. doi:10.1002/1521-3773(20010105)40:1<222::AID-ANIE222>3.0.CO;2-W |
| 25. | Nishiwaki, Y.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2002, 67, 5663–5668. doi:10.1021/jo025632d |
© 2013 Melone and Punta; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)