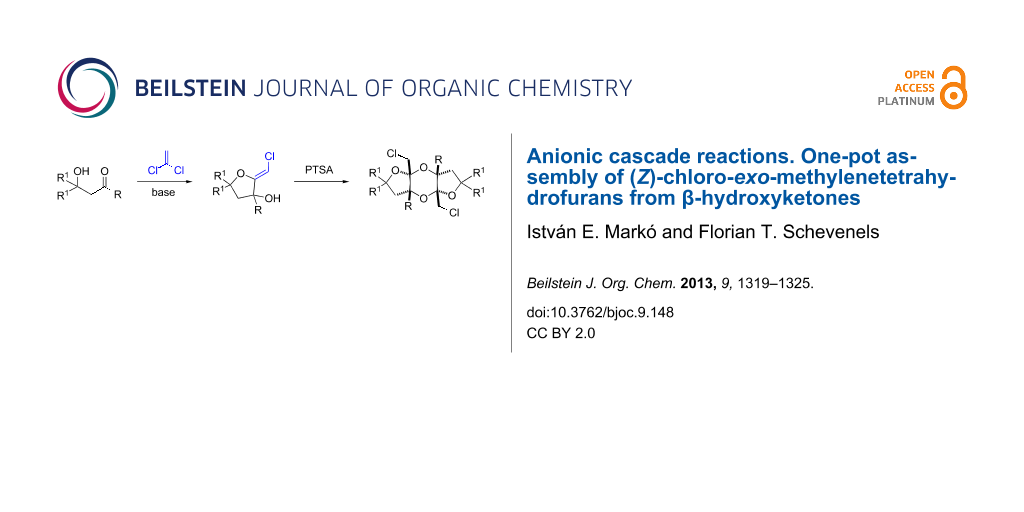
Abstract
The assembly of (Z)-chloro-exo-methylenetetrahydrofurans by an original and connective anionic cascade sequence is reported. Base-catalysed condensation of β-hydroxyketones with 1,1-dichloroethylene generates, in moderate to good yields, the corresponding (Z)-chloro-exo-methylenetetrahydrofurans. Acidic treatment of this motif leads to several unexpected dimers, possessing unique structural features.

Graphical Abstract
Introduction
Recently, we have shown that simple ketones reacted with 1,1-dichloroethylene, in the presence of potassium tert-butoxide, to afford rare (Z)-chloro-exo-methyleneketals [1-3]. This unique transformation is particularly efficient in the case of six-membered ring ketones (Scheme 1). In an attempt to extend the scope of this cascade process to acyclic ketones, acetone was submitted to the usual reaction conditions. To our surprise, the expected adduct 5 was formed in only poor yields. The major product was isolated in 36% yield and its structure was unambiguously determined as the (Z)-chloro-exo-methylenetetrahydrofuran 6.
![[1860-5397-9-148-i1]](/bjoc/content/inline/1860-5397-9-148-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of (Z)-chloro-exo-methyleneketals.
Scheme 1: Formation of (Z)-chloro-exo-methyleneketals.
The generation of 6 can be rationalized as depicted in Scheme 2. Under the basic conditions employed for the cascade reaction, 1,1-dichloroethylene is converted into the corresponding chloro-acetylene anion 9 [4,5]. This nucleophilic species adds rapidly, though reversibly, to acetone, leading ultimately to the formation of 5. However, in this case, competitive aldol reaction appears to take place, delivering the adduct 8. The subsequent addition of 9 then affords the intermediate 10, which undergoes a 5-exo-dig cyclisation, ultimately yielding the observed (Z)-chloro-exo-methylenetetrahydrofuran 6.
![[1860-5397-9-148-i2]](/bjoc/content/inline/1860-5397-9-148-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanism of formation of (Z)-chloro-exo-methylenetetrahydrofurans.
Scheme 2: Mechanism of formation of (Z)-chloro-exo-methylenetetrahydrofurans.
This structural motif constitutes the core of several biologically active compounds, including antimicrobial agents, fungicides and interesting competitive inhibitors of S-adenosyl-L-homocysteine (AdoHcy) hydrolase, one of the target enzymes extensively studied in the context of antiviral chemotherapy [6-9]. Despite their utility, only a limited number of multistep methods have been described for the preparation of this subunit [10-16]. The paucity of efficient synthetic approaches towards this family of compounds and the desire to assess the scope and limitations of this method prompted us to further investigate this transformation.
Results and Discussion
When 4-hydroxy-2-butanone (11) was treated with lithium chloroacetylide, generated in situ from dichloroethylene and LDA, the diol 12 was obtained in 77% yield. This adduct smoothly cyclized to 13 upon addition of sodium or potassium tert-butoxide, thereby supporting our mechanistic proposal. Repeating the process without isolation of the intermediate diol 12 proved to be even more efficient, affording the (Z)-chloro-exo-methylenetetrahydrofuran 13 in 66% overall yield (Scheme 3).
![[1860-5397-9-148-i3]](/bjoc/content/inline/1860-5397-9-148-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Stepwise formation of (Z)-chloro-exo-methylenetetrahydrofurans.
Scheme 3: Stepwise formation of (Z)-chloro-exo-methylenetetrahydrofurans.
It is interesting to note that, in the case of the substrate 11, the adduct 13 could be isolated in an improved 81% yield when potassium tert-butoxide was employed as the sole base (Scheme 4). In stark contrast, applying this procedure to the keto-alcohol 14 resulted in a mediocre 13% yield of 6, probably due to a rapid retro-aldol reaction of the derived, rather hindered, potassium alkoxide. Applying the LDA/KOt-Bu protocol improved the yield to 41%.
![[1860-5397-9-148-i4]](/bjoc/content/inline/1860-5397-9-148-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Optimized protocols to form (Z)-chloro-exo-methylenetetrahydrofurans.
Scheme 4: Optimized protocols to form (Z)-chloro-exo-methylenetetrahydrofurans.
It thus transpires that good to excellent yields of (Z)-chloro-exo-methylenetetrahydrofurans could be obtained by the judicious choice of the bases employed to promote this cascade process. Having delineated some suitable conditions, the scope and limitations of this methodology were investigated [17-27]. Some selected examples are collected in Table 1.
Table 1: Preparation of (Z)-chloro-exo-methylenetetrahydrofurans.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-148-i12.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yielda |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-148-i13.svg?max-width=637&scale=1.0)
17 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-148-i14.svg?max-width=637&scale=1.0)
25 |
39%b |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-148-i15.svg?max-width=637&scale=1.0)
18 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-148-i16.svg?max-width=637&scale=1.0)
26 |
80%c |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-148-i17.svg?max-width=637&scale=1.0)
19 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-148-i18.svg?max-width=637&scale=1.0)
27 |
73%c |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-148-i19.svg?max-width=637&scale=1.0)
20 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-148-i20.svg?max-width=637&scale=1.0)
28 |
38%c
47%b |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-148-i21.svg?max-width=637&scale=1.0)
21 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-148-i22.svg?max-width=637&scale=1.0)
29 |
40%b |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-148-i23.svg?max-width=637&scale=1.0)
22 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-148-i24.svg?max-width=637&scale=1.0)
30 |
42%c |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-148-i25.svg?max-width=637&scale=1.0)
23 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-148-i26.svg?max-width=637&scale=1.0)
31 |
77%c |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-148-i27.svg?max-width=637&scale=1.0)
24 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-148-i28.svg?max-width=637&scale=1.0)
32 |
10%c
61%b |
aAll yields are for pure, isolated products. bIn these cases, 2.2 equiv of C2H2Cl2 and 4.4 equiv of LDA were used in the first step, followed by 1 equiv of t-BuOK in the second step. cFor these substrates, 2.5 equiv of C2H2Cl2 and 5 equiv of t-BuOK were used to generate the acetylide.
As can be seen from Table 1, the reaction proves to be quite general and high-yielding for several primary alcohols (Table 1, entries 2, 3, 7 and 8). The yield was somewhat lower for two of them (Table 1, entries 4 and 6) due to rapid retro-aldol reactions and to enhanced steric hindrance, respectively. In the case of tertiary alcohols, the reaction proceeded with acceptable yields, especially in view of the one-pot nature of our procedure (Table 1, entries 1 and 5).
Having access to a broad range of substituted (Z)-chloro-exo-methylenetetrahydrofurans, a brief survey of their reactivity was performed. Several reactions involving the vinyl chloride function proved unsuccessful [28,29]. Attempts to perform oxidative rearrangement and dehydration failed and functionalisation of the hydroxy group appeared difficult [30,31]. Initially, the adduct 13 was treated with aqueous hydrochloric acid, in the hope of generating the corresponding dihydrofuran carbaldehyde 33. Instead, the diol 34 was obtained in 52% yield as a 1:1 mixture of diastereoisomers. It thus transpires that protonation and hydration of the exo-methylene double bond of 13 proceeded faster than the expected rearrangement of the tertiary allylic alcohol (Scheme 5).
![[1860-5397-9-148-i5]](/bjoc/content/inline/1860-5397-9-148-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Hydration of (Z)-chloro-exo-methylenetetrahydrofurans.
Scheme 5: Hydration of (Z)-chloro-exo-methylenetetrahydrofurans.
To promote such an acid-catalyzed rearrangement, the (Z)-chloro-exo-methylenetetrahydrofuran 13 was treated with a catalytic amount of para-toluenesulfonic acid. To our surprise, the anti-dioxane 35 and its syn-derivative 36 were obtained as a 1:5 mixture of diastereoisomers that could be separated (Scheme 6). Their structure was unambiguously established by single-crystal X-ray diffraction analysis, as shown in Figure 1. Increasing the steric hindrance at the tertiary alcohol site resulted in the exclusive formation of the syn-dioxanes 37 and 38 respectively, albeit at the expense of the yield [32-38].
![[1860-5397-9-148-i6]](/bjoc/content/inline/1860-5397-9-148-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-148-1]](/bjoc/content/figures/1860-5397-9-148-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray diffraction analysis of dioxanes 35 and 36.
Figure 1: X-ray diffraction analysis of dioxanes 35 and 36.
When the dimethyl adduct 6 was submitted to the same conditions, a new product [39-45] 41 was formed besides the dioxanes 39 and 40 (Scheme 7). Its structure was unambiguously established as the spirocyclic dimer 41 by single-crystal X-ray diffraction analysis. It is noteworthy that a single diastereoisomer was generated in this transformation.
![[1860-5397-9-148-i7]](/bjoc/content/inline/1860-5397-9-148-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Formation of a new spirocyclic dimer.
Scheme 7: Formation of a new spirocyclic dimer.
The formation of these unique compounds can be rationalized as depicted in Scheme 8. Under acidic conditions, the (Z)-chloro-exo-methylenetetrahydrofuran 42 is converted into the oxonium cation 43. Subsequent capture of this intermediate 43 by a second molecule of 42 can occur via two different pathways. The first one involves the addition of the enol ether function of 42 onto 43, leading to the creation of a new C–C bond with concomitant generation of another oxonium species 47. Intramolecular capture of this electrophile by the pendant hydroxy substituent then delivers the spirocyclic adduct 48. Alternatively, reaction of the tertiary alcohol of 42 with the oxonium cation 43 affords the ketal 44. Protonation of the enol ether function, followed by 6-exo-trig cyclization, completes this sequence of events and provides the dioxane derivative 46. Interestingly, no reaction was observed when the spirocycle 41 and the dioxanes 35, 36 and 39 were reacted under acidic conditions, indicating that the final step is not reversible.
![[1860-5397-9-148-i8]](/bjoc/content/inline/1860-5397-9-148-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Mechanism leading to dioxanes and spirocycles.
Scheme 8: Mechanism leading to dioxanes and spirocycles.
It is noteworthy that the dioxanes possess predominantly (13) or exclusively (26 and 27) the syn–syn relative stereochemistry at the ring junctions. This selectivity can be rationalized by an initial addition of the nucleophile syn to the alcohol function, as shown in Scheme 9. Interestingly, the homochiral (S,S;R,R)-dimer, leading to the syn–syn product, is preferred over the heterochiral (S,R;R,S)-adduct, affording the syn–anti isomer. Increasing the steric hindrance at the tertiary alcohol center of 49 leads to prohibitive steric repulsion during the heterodimer formation. A similar facial selectivity can explain the formation of 41 as a single diastereoisomer.
![[1860-5397-9-148-i9]](/bjoc/content/inline/1860-5397-9-148-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: (S,S)-syn and (S,R)-syn approaches.
Scheme 9: (S,S)-syn and (S,R)-syn approaches.
When the adduct 25, bearing a gem-dimethyl substituent was submitted to this transformation, no dioxane was observed. The major product proved to be the spirocyclic adduct 53, accompanied by the bridged bis-ketal 52, in 41% and 13% yield respectively (Scheme 10). Treatment of the isopropyl derivative 29 under the same acidic conditions provided the triene 54 in 57% yield. Addition of aqueous HCl generated the hemi-ketal 55 in 43% yield. The starting material was recovered in 52% yield.
![[1860-5397-9-148-i10]](/bjoc/content/inline/1860-5397-9-148-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Formation of a bridged dimer and a triene.
Scheme 10: Formation of a bridged dimer and a triene.
The structures of compounds 53 and 55 were unambiguously established by single-crystal X-ray diffraction analysis (Figure 2).
![[1860-5397-9-148-2]](/bjoc/content/figures/1860-5397-9-148-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray diffraction analysis of two new dimers.
Figure 2: X-ray diffraction analysis of two new dimers.
Once again, increasing the steric effect around the tertiary alcohol function of the (Z)-chloro-exo-methylenetetrahydrofurans has a profound influence on the fate of the condensation reaction. A plausible mechanistic rationale is provided in Scheme 11.
![[1860-5397-9-148-i11]](/bjoc/content/inline/1860-5397-9-148-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Mechanism leading to bridged and dienic dimers.
Scheme 11: Mechanism leading to bridged and dienic dimers.
In the presence of the gem-dimethyl substituent, dehydration of 42 to afford 56 appears to proceed rapidly, probably owing to a release of steric strain. The loss of water occurs especially readily when R’ = Et or iPr and to a lesser extent when R’ = Me, indicating that steric decompression may indeed be operational in these cases. Protonation of diene 56 leads to the oxonium ion 57, which undergoes a 1,4-addition of another 56 unit, affording the new cation 58. At this stage, two different pathways can be followed. Either carbocation 58 can lose a proton, generating the observed triene 59, or it may undergo addition of a water molecule, affording the hemiketal 60. Addition of a proton to the vinyl ether function, followed by intramolecular capture by the hydroxy group, finally provides the unique bridged adduct 62.
Conclusion
In summary, a unique anionic cascade process, leading to the efficient and connective assembly of (Z)-chloro-exo-methylenetetrahydrofurans from β-hydroxyketones, has been uncovered and developed. The reactivity of this unusual motif has been briefly investigated, and dimers, possessing rather unusual structures, have been obtained. It is noteworthy that some of these dimeric products form the core of interesting biologically active compounds and of unique natural products.
Experimental
General procedure for the synthesis of chloromethylenefurans
To 40 mL of anhydrous THF, cooled to 0 °C, 1.2 mL (15.0 mmol, 2.5 equiv) of dichloroethylene and 3.37 g of potassium tert-butoxide were added. After 15 minutes, 700 mg (6.0 mmol, 1 equiv) of hydroxyketone in 1 mL of THF were added. After one hour, 20 mL of water was added and the mixture was neutralized with diluted sulfuric acid. The aqueous layer was extracted with 2 × 20 mL of DCM. The organic layer was dried, filtered and concentrated. The crude product was further purified by chromatography over silica gel providing 820 mg (4.6 mmol, 77%) of chloromethylenefuran 31 as a white solid. For full details, see Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental procedures and analytical data of cited compounds. | ||
| Format: PDF | Size: 988.7 KB | Download |
References
-
Schevenels, F.; Markó, I. E. Chem. Commun. 2011, 47, 3287. doi:10.1039/c0cc05186a
Return to citation in text: [1] -
Schevenels, F.; Markó, I. E. Org. Lett. 2012, 14, 1298. doi:10.1021/ol300186s
Return to citation in text: [1] -
Schevenels, F.; Tinant, B.; Declercq, J.-P.; Markó, I. E. Chem.–Eur. J. 2013, 19, 4335. doi:10.1002/chem.201201505
Return to citation in text: [1] -
Viehe, H. G. Chem. Ber. 1959, 92, 1270. doi:10.1002/cber.19590920607
Return to citation in text: [1] -
Kloster-Jenson, E. Tetrahedron 1971, 27, 33. doi:10.1016/S0040-4020(01)92395-6
Return to citation in text: [1] -
Prakash, N. J.; Davis, G. F.; Jarvi, E. T.; Edwards, M. L.; McCarthy, J. R.; Bowlin, T. L. Life Sci. 1992, 50, 1425. doi:10.1016/0024-3205(92)90261-M
Return to citation in text: [1] -
Buchecker, R.; Fuenfschilling, J.; Marck, G. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 1997, 302, 295. doi:10.1080/10587259708041840
Return to citation in text: [1] -
Kumamoto, H.; Onuma, S.; Tanaka, H.; Kitade, Y. Nucleic Acids Symp. Ser. 2003, 3, 13. doi:10.1093/nass/3.1.13
Return to citation in text: [1] -
Jarvi, E. T.; McCarthy, J. R.; Mehdi, S.; Matthews, D. P.; Edwards, M. L.; Prakash, N. J.; Bowlin, T. L.; Sunkara, P. S.; Bey, P. J. Med. Chem. 1991, 34, 647. doi:10.1021/jm00106a028
Return to citation in text: [1] -
Kumamoto, H.; Onuma, S.; Tanaka, H. J. Org. Chem. 2004, 69, 72. doi:10.1021/jo030256y
Return to citation in text: [1] -
Wnuk, S. F.; Dalley, N. K.; Robins, M. J. J. Org. Chem. 1993, 58, 111. doi:10.1021/jo00053a022
Return to citation in text: [1] -
Xu, M.; Miao, Z.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2005, 88, 2918. doi:10.1002/hlca.200590235
Return to citation in text: [1] -
Csuk, R.; Glänzer, B. Tetrahedron 1991, 47, 1655. doi:10.1016/S0040-4020(01)96909-1
Return to citation in text: [1] -
Gómez, A. M.; Danelón, G. O.; Pedregosa, A.; Valverde, S.; López, J. C. Chem. Commun. 2002, 2024. doi:10.1039/b205121d
Return to citation in text: [1] -
Blandino, M.; McNelis, E. Org. Lett. 2002, 4, 3387. doi:10.1021/ol026422q
Return to citation in text: [1] -
Craig, G. W.; Sternberg, E. D.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1986, 51, 1258. doi:10.1021/jo00358a017
Return to citation in text: [1] -
Wasserman, H. H.; Hearn, M. J.; Cochoy, R. E. J. Org. Chem. 1980, 45, 2874. doi:10.1021/jo01302a023
Return to citation in text: [1] -
Hitchcock, S. R.; Perron, F.; Martin, V. A.; Albizati, K. F. Synthesis 1990, 1059. doi:10.1055/s-1990-27093
Return to citation in text: [1] -
Einhorn, J.; Soulier, J.-L.; Bacquet, C.; Lelandais, D. Can. J. Chem. 1983, 61, 584. doi:10.1139/v83-104
Return to citation in text: [1] -
Ueki, T.; Doe, M.; Morimoto, Y.; Kinoshita, T.; Tanaka, R.; Yoshihara, K. J. Heterocycl. Chem. 2001, 38, 165. doi:10.1002/jhet.5570380124
Return to citation in text: [1] -
O’Neil, G. W.; Miller, M. M.; Carter, K. P. Org. Lett. 2010, 12, 5350. doi:10.1021/ol1024635
Return to citation in text: [1] -
Kim, K. S.; Song, Y. H.; Lee, N. H.; Hahn, C. S. Tetrahedron Lett. 1986, 27, 2875. doi:10.1016/S0040-4039(00)84667-5
Return to citation in text: [1] -
Schneider, C.; Hansch, M.; Weide, T. Chem.–Eur. J. 2006, 11, 3010. doi:10.1002/chem.200400951
Return to citation in text: [1] -
Lin, K.-W.; Tsai, C.-H.; Hsieh, I.-L.; Yan, T.-H. Org. Lett. 2008, 10, 1927. doi:10.1021/ol8004326
Return to citation in text: [1] -
Sulmon, P.; De Kimpe, N.; Schamp, N. Org. Prep. Proced. Int. 1989, 21, 91. doi:10.1080/00304948909356351
Return to citation in text: [1] -
Trost, B. M.; Yang, H.; Thiel, O. R.; Frontier, A. J.; Brindle, C. S. J. Am. Chem. Soc. 2007, 129, 2206. doi:10.1021/ja067305j
Return to citation in text: [1] -
Gerwick, B. C., III; Fields, S. C.; Graupner, P. R.; Schmitzer, P. R.; Brewster, W. K. Methylidene Mevalonates and their Use as Herbicides. U.S. Patent CA2527439A1, May 19, 2006.
Return to citation in text: [1] -
Scheiper, B.; Bonnekessel, M.; Krause, H.; Fürstner, A. J. Org. Chem. 2004, 69, 3943. doi:10.1021/jo0498866
Return to citation in text: [1] -
Qing, F.-L.; Wang, R.; Li, B.; Zheng, X.; Meng, W.-D. J. Fluorine Chem. 2003, 120, 21. doi:10.1016/S0022-1139(02)00279-8
Return to citation in text: [1] -
Dauben, W. G.; Michno, D. M. J. Org. Chem. 1977, 42, 682. doi:10.1021/jo00424a023
Return to citation in text: [1] -
Ishihara, K.; Kubota, M.; Kurihara, H.; Yamamoto, H. J. Am. Chem. Soc. 1995, 117, 4413. doi:10.1021/ja00120a030
Return to citation in text: [1] -
Yates, P.; Burke, P. M. Can. J. Chem. 1987, 65, 1695. doi:10.1139/v87-284
Return to citation in text: [1] -
Martin, O. R.; Rao, S. P.; Hendricks, C. A. V.; Mahnken, R. E. Carbohydr. Res. 1990, 202, 49. doi:10.1016/0008-6215(90)84070-B
Return to citation in text: [1] -
Adam, W.; Peters, C.; Sauter, M. Synthesis 1994, 111. doi:10.1055/s-1994-25416
Return to citation in text: [1] -
Dawid, M.; Warkentin, J. Can. J. Chem. 2003, 81, 598. doi:10.1139/v02-200
Return to citation in text: [1] -
Timmer, M. S. M.; Stocker, B. L.; Seeberger, P. H. J. Org. Chem. 2006, 71, 8294. doi:10.1021/jo061607m
Return to citation in text: [1] -
Mukherjee, S.; Roy, B. G.; Das, S. N.; Mandal, S. B. Tetrahedron Lett. 2012, 53, 4929. doi:10.1016/j.tetlet.2012.06.121
Return to citation in text: [1] -
Ting, C.-M.; Wang, C.-D.; Chaudhuri, R.; Liu, R.-S. Org. Lett. 2011, 13, 1702. doi:10.1021/ol2002144
Return to citation in text: [1] -
Chang, S.; Britton, R. Org. Lett. 2012, 14, 5844. doi:10.1021/ol302694s
Return to citation in text: [1] -
Tlais, S. F.; Dudley, G. B. Beilstein J. Org. Chem. 2012, 8, 1287. doi:10.3762/bjoc.8.146
Return to citation in text: [1] -
Brimble, M. A.; Finch, O. C.; Heapy, A. M.; Fraser, J. D.; Furkert, D. P.; O’Connor, P. D. Tetrahedron 2011, 67, 995. doi:10.1016/j.tet.2010.11.107
Return to citation in text: [1] -
Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698. doi:10.1021/ol102201z
Return to citation in text: [1] -
Fernandes, R. A.; Ingle, A. B. Synlett 2010, 158. doi:10.1055/s-0029-1218538
Return to citation in text: [1] -
Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842. doi:10.1021/jo802539z
Return to citation in text: [1] -
Seibert, S. F.; Krick, A.; Eguereva, E.; Kehraus, S.; Koenig, G. M. Org. Lett. 2007, 9, 239. doi:10.1021/ol0626802
Return to citation in text: [1]
| 1. | Schevenels, F.; Markó, I. E. Chem. Commun. 2011, 47, 3287. doi:10.1039/c0cc05186a |
| 2. | Schevenels, F.; Markó, I. E. Org. Lett. 2012, 14, 1298. doi:10.1021/ol300186s |
| 3. | Schevenels, F.; Tinant, B.; Declercq, J.-P.; Markó, I. E. Chem.–Eur. J. 2013, 19, 4335. doi:10.1002/chem.201201505 |
| 17. | Wasserman, H. H.; Hearn, M. J.; Cochoy, R. E. J. Org. Chem. 1980, 45, 2874. doi:10.1021/jo01302a023 |
| 18. | Hitchcock, S. R.; Perron, F.; Martin, V. A.; Albizati, K. F. Synthesis 1990, 1059. doi:10.1055/s-1990-27093 |
| 19. | Einhorn, J.; Soulier, J.-L.; Bacquet, C.; Lelandais, D. Can. J. Chem. 1983, 61, 584. doi:10.1139/v83-104 |
| 20. | Ueki, T.; Doe, M.; Morimoto, Y.; Kinoshita, T.; Tanaka, R.; Yoshihara, K. J. Heterocycl. Chem. 2001, 38, 165. doi:10.1002/jhet.5570380124 |
| 21. | O’Neil, G. W.; Miller, M. M.; Carter, K. P. Org. Lett. 2010, 12, 5350. doi:10.1021/ol1024635 |
| 22. | Kim, K. S.; Song, Y. H.; Lee, N. H.; Hahn, C. S. Tetrahedron Lett. 1986, 27, 2875. doi:10.1016/S0040-4039(00)84667-5 |
| 23. | Schneider, C.; Hansch, M.; Weide, T. Chem.–Eur. J. 2006, 11, 3010. doi:10.1002/chem.200400951 |
| 24. | Lin, K.-W.; Tsai, C.-H.; Hsieh, I.-L.; Yan, T.-H. Org. Lett. 2008, 10, 1927. doi:10.1021/ol8004326 |
| 25. | Sulmon, P.; De Kimpe, N.; Schamp, N. Org. Prep. Proced. Int. 1989, 21, 91. doi:10.1080/00304948909356351 |
| 26. | Trost, B. M.; Yang, H.; Thiel, O. R.; Frontier, A. J.; Brindle, C. S. J. Am. Chem. Soc. 2007, 129, 2206. doi:10.1021/ja067305j |
| 27. | Gerwick, B. C., III; Fields, S. C.; Graupner, P. R.; Schmitzer, P. R.; Brewster, W. K. Methylidene Mevalonates and their Use as Herbicides. U.S. Patent CA2527439A1, May 19, 2006. |
| 10. | Kumamoto, H.; Onuma, S.; Tanaka, H. J. Org. Chem. 2004, 69, 72. doi:10.1021/jo030256y |
| 11. | Wnuk, S. F.; Dalley, N. K.; Robins, M. J. J. Org. Chem. 1993, 58, 111. doi:10.1021/jo00053a022 |
| 12. | Xu, M.; Miao, Z.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2005, 88, 2918. doi:10.1002/hlca.200590235 |
| 13. | Csuk, R.; Glänzer, B. Tetrahedron 1991, 47, 1655. doi:10.1016/S0040-4020(01)96909-1 |
| 14. | Gómez, A. M.; Danelón, G. O.; Pedregosa, A.; Valverde, S.; López, J. C. Chem. Commun. 2002, 2024. doi:10.1039/b205121d |
| 15. | Blandino, M.; McNelis, E. Org. Lett. 2002, 4, 3387. doi:10.1021/ol026422q |
| 16. | Craig, G. W.; Sternberg, E. D.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1986, 51, 1258. doi:10.1021/jo00358a017 |
| 6. | Prakash, N. J.; Davis, G. F.; Jarvi, E. T.; Edwards, M. L.; McCarthy, J. R.; Bowlin, T. L. Life Sci. 1992, 50, 1425. doi:10.1016/0024-3205(92)90261-M |
| 7. | Buchecker, R.; Fuenfschilling, J.; Marck, G. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 1997, 302, 295. doi:10.1080/10587259708041840 |
| 8. | Kumamoto, H.; Onuma, S.; Tanaka, H.; Kitade, Y. Nucleic Acids Symp. Ser. 2003, 3, 13. doi:10.1093/nass/3.1.13 |
| 9. | Jarvi, E. T.; McCarthy, J. R.; Mehdi, S.; Matthews, D. P.; Edwards, M. L.; Prakash, N. J.; Bowlin, T. L.; Sunkara, P. S.; Bey, P. J. Med. Chem. 1991, 34, 647. doi:10.1021/jm00106a028 |
| 4. | Viehe, H. G. Chem. Ber. 1959, 92, 1270. doi:10.1002/cber.19590920607 |
| 5. | Kloster-Jenson, E. Tetrahedron 1971, 27, 33. doi:10.1016/S0040-4020(01)92395-6 |
| 39. | Chang, S.; Britton, R. Org. Lett. 2012, 14, 5844. doi:10.1021/ol302694s |
| 40. | Tlais, S. F.; Dudley, G. B. Beilstein J. Org. Chem. 2012, 8, 1287. doi:10.3762/bjoc.8.146 |
| 41. | Brimble, M. A.; Finch, O. C.; Heapy, A. M.; Fraser, J. D.; Furkert, D. P.; O’Connor, P. D. Tetrahedron 2011, 67, 995. doi:10.1016/j.tet.2010.11.107 |
| 42. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698. doi:10.1021/ol102201z |
| 43. | Fernandes, R. A.; Ingle, A. B. Synlett 2010, 158. doi:10.1055/s-0029-1218538 |
| 44. | Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842. doi:10.1021/jo802539z |
| 45. | Seibert, S. F.; Krick, A.; Eguereva, E.; Kehraus, S.; Koenig, G. M. Org. Lett. 2007, 9, 239. doi:10.1021/ol0626802 |
| 32. | Yates, P.; Burke, P. M. Can. J. Chem. 1987, 65, 1695. doi:10.1139/v87-284 |
| 33. | Martin, O. R.; Rao, S. P.; Hendricks, C. A. V.; Mahnken, R. E. Carbohydr. Res. 1990, 202, 49. doi:10.1016/0008-6215(90)84070-B |
| 34. | Adam, W.; Peters, C.; Sauter, M. Synthesis 1994, 111. doi:10.1055/s-1994-25416 |
| 35. | Dawid, M.; Warkentin, J. Can. J. Chem. 2003, 81, 598. doi:10.1139/v02-200 |
| 36. | Timmer, M. S. M.; Stocker, B. L.; Seeberger, P. H. J. Org. Chem. 2006, 71, 8294. doi:10.1021/jo061607m |
| 37. | Mukherjee, S.; Roy, B. G.; Das, S. N.; Mandal, S. B. Tetrahedron Lett. 2012, 53, 4929. doi:10.1016/j.tetlet.2012.06.121 |
| 38. | Ting, C.-M.; Wang, C.-D.; Chaudhuri, R.; Liu, R.-S. Org. Lett. 2011, 13, 1702. doi:10.1021/ol2002144 |
| 30. | Dauben, W. G.; Michno, D. M. J. Org. Chem. 1977, 42, 682. doi:10.1021/jo00424a023 |
| 31. | Ishihara, K.; Kubota, M.; Kurihara, H.; Yamamoto, H. J. Am. Chem. Soc. 1995, 117, 4413. doi:10.1021/ja00120a030 |
| 28. | Scheiper, B.; Bonnekessel, M.; Krause, H.; Fürstner, A. J. Org. Chem. 2004, 69, 3943. doi:10.1021/jo0498866 |
| 29. | Qing, F.-L.; Wang, R.; Li, B.; Zheng, X.; Meng, W.-D. J. Fluorine Chem. 2003, 120, 21. doi:10.1016/S0022-1139(02)00279-8 |
© 2013 Markó and Schevenels; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)