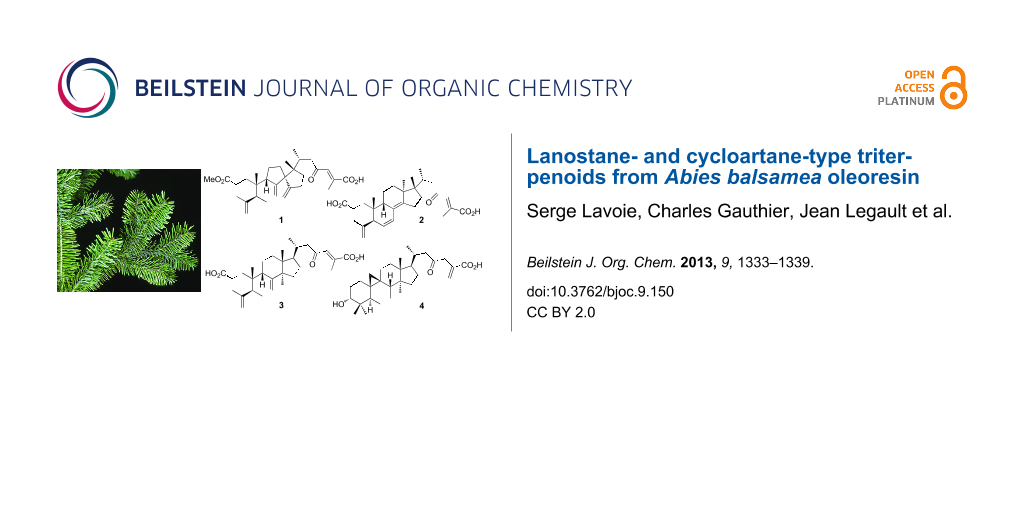
Lanostane- and cycloartane-type triterpenoids from Abies balsamea oleoresin
‡,1,
‡,1,2,
1,
1,
1 and
1
Serge Lavoie
Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
Université de Poitiers, Institut de Chimie IC2MP, UMR-CNRS 7285, 4 rue Michel Brunet, 86022 Poitiers, France
Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
1Université du Québec à Chicoutimi, Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale, Laboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, 555 boul. de l'Université, Chicoutimi (Québec) G7H 2B1, Canada
2Université de Poitiers, Institut de Chimie IC2MP, UMR-CNRS 7285, 4 rue Michel Brunet, 86022 Poitiers, France
Corresponding author email
‡ Equal contributors
Associate Editor: A. Kirschning Beilstein J. Org. Chem.2013,9, 1333–1339.https://doi.org/10.3762/bjoc.9.150 Received 15 Apr 2013,
Accepted 12 Jun 2013,
Published 04 Jul 2013
Phytochemical analysis of A. balsamea oleoresin led to the isolation of three new 3,4-seco-lanostane triterpenoids 1–3, one new cycloartane triterpenoid 4 along with fourteen known terpenoids. Structure determinations were based on extensive 1D/2D NMR, IR and MS spectroscopic analyses, and comparison with literature data. The isolated compounds were evaluated in vitro for their cytotoxicity against human cell lines (A549, DLD-1, WS1) and their antibacterial activity against E. coli and S. aureus. Abiesonic acid (6) exhibited weak cytotoxic activity against A549 (IC50 = 22 µM) while compounds 1 and 4 were weakly active against S. aureus (MIC = 25 µM).
The genus Abies (Pinaceae) comprises 46 species of evergreen conifers [1]. Most of them are found in temperate and boreal regions of the northern hemisphere. The first phytochemical investigation of Abies species was undertaken 75 years ago by Takahashi [2]. Since then, more than 277 secondary metabolites have been isolated, and mainly identified as terpenoids, flavonoids and lignans [3]. Balsam fir Abies balsamea (L.) Mill., a popular Christmas tree in Canada, has been used traditionally by North American aboriginal people as an antiseptic, tuberculosis remedy, and venereal aid [4]. In recent years, we have become interested in studying the bioactive constituents of A. balsamea. Our work allowed the identification of antibacterial sesquiterpenoids, active against S. aureus, from balsam fir essential oil [5]. We also isolated two cytotoxic tetraterpenoids from the cortical oleoresin of the tree bark, featuring an unprecedented C40 scaffold [6]. Herein, we describe the further phytochemical study of A. balsamea oleoresin, which led to the isolation and structure elucidation of three 3,4-seco-lanostane-type triterpenoids 1–3, one cycloartane-type triterpenoid 4 and fourteen known terpenoids. The antibacterial (E. coli and S. aureus) and cytotoxic (A549, DLD-1 and WS1) activities of the isolated compounds are also reported.
Results and Discussion
The oleoresin of A. balsamea (1st lot) was fractionated by silica gel column chromatography with hexanes/EtOAc (100:0 → 93:7) and MeOH as eluent. Both hexanes/EtOAc 93:7 and MeOH fractions were combined and concentrated under reduced pressure. Purification of this extract using a combination of silica gel or polyamide column chromatography and reversed phase C18 HPLC resulted in the isolation of three new (1–3) and six known terpenoids (Figure 1). In another experiment, oleoresin (2nd lot) was triturated with hexanes. The precipitate was subjected to successive silica gel column chromatography followed by reversed phase C18 HPLC to give one new (4) as well as three known terpenoids. Similarly, purification of the filtrate afforded five known terpenoids. Based on their spectroscopic data (IR, MS and NMR) and comparison with literature values, the structures of the known compounds were elucidated as awashishinic acid (5) [7], abiesonic acid (6) [6], firmanoic acid (7) [8], (22Z)-3,4-seco-9βH-lanosta-4(28),7,22,24-tetraen-23,26-olid-3-oic acid (8) [9], (25R)-3,4-seco-9βH-lanosta-4(28),7-diene-3,26-dioic acid (9) [10], abiesolidic acid (10) [10,11], (23R,25R)-3,4-seco-17,14-friedo-9βH-lanosta-4(28),6,8(14)-trien-26,23-olid-3-oic acid (11) [10], (24E)-3,4-seco-9βH-lanosta-4(28),7,24-triene-3,26-dioic acid (12) [12], abiesanordine C (13) [13], methyl 13-oxo-podocarp-8(14)-en-15-oate (14) [14], 15-hydroxydehydroabietic acid (15) [15], methyl 15-hydroxydehydroabietate (16) [16], (12E)-8-hydroxy-15-nor-12-labden-14-al (17) [17] and 8-hydroxy-14,15-dinor-11-labden-13-one (18) [13,18] (Figure 1). 1H and 13C NMR spectroscopic data of known compounds (5–18) are given in Supporting Information File 1.
Compound 1 was isolated as a white amorphous powder. Its molecular formula was established as C31H44O5 from the [M + H]+ peak at m/z 497.3261 (calcd 497.3262) in the positive HRESIMS, indicating ten degrees of unsaturation. The IR spectrum displayed strong absorption bands at 1692 and 1736 cm−1 indicative of carboxylic acid functionalities. The 13C NMR and DEPT spectroscopic data (Table 1) exhibited 31 carbons including one carbonyl carbon at δC 202.4, and two carboxylic carbons at δC 172.4 and 174.8. The 1H NMR data (Table 2) exhibited six olefinic signals at δH 4.73 (s), 4.77 (s), 4.78 (s), 4.86 (s), 5.48 (dd, J = 6.2, 3.1 Hz) and 7.11 (br s), one methoxy methyl at δH 3.67 (s), four tertiary methyl at δH 0.90 (s), 0.92 (s), 1.75 (s) and 2.18 (s) and one secondary methyl at δH 0.85 (d, J = 6.4 Hz). Detail analysis of the above NMR information, together with 1H–1H COSY, HSQC and HMBC analyses indicated that 1 shares the same structure with abiesonic acid (6), previously isolated from A. balsamea[6], but with an additional methoxy group. An HMBC cross-peak between this methyl signal and the carbon at δC 174.8 (C-3) allowed the assignment of compound 1 as (−)-rel-abiesonic acid 3-methyl ester.
Table 1:13C NMR spectroscopic data (100 MHz, CDCl3) of compounds 1–4.
Position
1
2
3
4
1
30.5
28.3
28.8
27.5
2
29.2
29.8
29.2
28.5
3
174.8
181.6
180.8
77.1
4
149.2
145.6
149.7
39.5
5
44.0
50.6
45.3
41.1
6
30.9
127.0
29.7
21.1
7
122.4
125.2
118.0
25.6
8
143.4
125.4
146.3
48.0
9
49.5
39.4
38.6
19.7
10
36.9
37.0
36.3
26.5
11
22.5
19.6
18.5
26.2
12
31.2
32.0
33.8
32.8
13
63.5
47.4
43.8
45.4
14
160.9
146.2
51.7
49.0
15
27.8
23.9
34.0
35.4
16
36.1
36.3
28.5
28.3
17
50.2
49.1
53.1
52.2
18
17.7
21.9
21.7
18.1
19
24.7
21.8
24.1
29.8
20
33.8
35.1
33.3
32.9
21
16.4
15.9
19.5
19.3
22
48.3
48.9
51.9
50.0
23
202.4
202.5
202.4
207.6
24
134.9
133.0
134.4
46.1
25
138.7
140.4
139.3
133.9
26
172.4
173.4
172.8
170.8
27
14.0
14.0
13.9
130.5
28
111.9
115.6
112.0
25.8
29
26.1
24.8
26.0
21.2
30
106.9
15.8
27.5
19.3
OMe
51.7
–
–
–
Table 2:1H NMR spectroscopic data (400 MHz, CDCl3) of compounds 1–4.
Position
δH (J in Hz)
1
2
3
4
1
1.74, m, 1.62, m
1.60, m
1.73, m, 1.60, m
1.85, m, 1.01, m
2
2.30, m
2.31, m
2.32, m
1.93, m, 1.64, m
3
–
–
–
3.48, t (2.4)
5
2.08, m
2.63, d (5.4)
2.08, m
1.82, m
6
2.40, m, 2.13, m
5.39, dd (9.9, 5.5)
2.27, m, 1.99, m
1.48, m, 0.77, m
7
5.48, dd (6.2, 3.1)
6.22, d (10.0)
5.33, br s
1.30, m, 1.11, m
8
–
–
–
1.54, m
9
2.06, m
2.43, m
2.59, m
–
11
1.59, m, 1.40, m
1.62, m
1.60, m
2.00, m, 1.13, m
12
1.77, m, 1.32, m
1.65, m
1.83, m, 1.67, m
1.62, m
15
2.48, m, 2.37, m
2.41, m, 2.32, m
1.52, m
1.31, m
16
1.55, m
1.73, m, 1.54, m
1.92, m, 1.26, m
1.87, m, 1.27, m
17
–
–
1.54, m
1.61, m
18
0.90, s
1.16, s
0.80, s
1.00, s
19
0.92, s
0.87, s
0.86, s
0.52, d (3.9), 0.35, d (3.9)
20
2.39, m
2.24, m
2.03, m
2.02, m
21
0.85, d (6.4)
0.80, d (6.5)
0.91, d (6.2)
0.88, d (6.8)
22
2.49, m, 2.25, m
2.85, m 2.16, br d (12.3)
2.64, m 2.32, m
2.56, dd (16.0, 2.1), 2.24, dd (16.1, 10.2)
24
7.11, br s
7.23, br s
7.15, s
3.42, d (17.0) 3.36, d (17.1)
27
2.18, s
2.22, d (1.0)
2.21, s
6.45, br s 5.73, br s
28
4.86, s, 4.78, s
4.98, br s, 4.76, d (2.4)
4.88, s, 4.82, s
0.95, s
29
1.75, s
1.79, s
1.80, s
0.88, s
30
4.77, s, 4.73, s
0.69, s
1.04, s
0.90, s
OMe
3.67, s
–
–
–
Compound 2, obtained as a white amorphous powder, possessed a molecular formula of C30H42O4 with ten degrees of unsaturation based on the [M + H]+ peak at m/z 483.3087 (calcd 483.3105) in the positive HRESIMS. The IR absorption bands showed the presence of carboxylic acid (1702 cm−1) and olefin (1635 cm−1) functionalities. The 13C NMR spectroscopic data of 2 (Table 1) displayed 30 carbon signals, which by the assistance of a DEPT experiment, were identified as six methyl, seven sp3 methylene and three sp3 methine groups, three sp3 quaternary carbon atoms, one sp2 methylene and three sp2 methine groups, and seven sp2 quaternary carbon atoms. A 1H–1H COSY experiment provided correlations from H2-1 to H2-2, H-6 to H-5 and H-7, H2-11 to H-9 and H2-12, H2-15 to H2-16 and H-20 to H3-21 and H2-22 (Figure 2). Analysis of HMBC spectra indicated correlations from H3-19 to C-1, C-5, C-9 and C-10; from H3-29 to C-4, C-5 and C-28; from H-7 to C-8; from H3-18 to C-12, C-13, C-14 and C-17; from H3-30 to C-13, C-16, C-17 and C-20; from H3-21 to C-17, C-20 and C-22; from H2-22 and H-24 to C-23; and from H3-27 to C-24, C-25 and C-26. The relative configuration of 2 was determined by analysis of a NOESY experiment, which provided correlations (Figure 2) of H-5 to H2-2; H-28Z to H-9; H-22a (δH 2.85) to H3-18 and H3-21; H3-18 to H-22b (δH 2.16) and H-24; H-24 to H-20 and H-22b. These correlations indicated the α-orientation of H-5 and H3-30 and the β-orientation of H-9, H3-18 and H3-19. All these facts suggested that compound 2 was strongly similar to cis-sibiric acid [19]. Since the chemical shift of H-24 in cis-sibiric acid (δH 6.15) was upfield of the signal for 1 (δH 7.11), 2 (δH 7.23), 6 (δH 7.13) and 7 (δH 7.07), this suggested that the trans-stereoisomer was isolated instead of the cis-one (See Table 2 and Supporting Information File 1). This was further confirmed by NOESY correlation of H-24 to H-20 and H3-30, but not to H3-27. Consequently, the structure of 2 was determined as (−)-rel-(24E)-23-oxo-3,4-seco-9βH-lanosta-4(28),6,8(14),24-tetraen-3,26-dioic acid.
Figure 2:
Selected COSY (▬), HMBC (blue arrows) and NOESY (red arrows) correlations for compounds 2–4.
Figure 2:
Selected COSY (▬), HMBC (blue arrows) and NOESY (red arrows) correlations for compounds 2–4.
Compound 3, a white amorphous powder, possessed a molecular formula of C30H44O5 based on the [M + H]+ peak at m/z 485.3250 (calcd 485.3262) in the positive HRESIMS, suggesting nine degrees of unsaturation. The IR spectrum implied the existence of carboxylic acid (1703 cm−1) and olefin (1633 cm−1) functionalities. The 13C NMR spectroscopic data of 3 resembled those of (24E)-3,4-seco-9βH-lanosta-4(28),7,24-triene-3,26-dioic acid (12) [12] except for change at δC 33.3 (C-20), 19.5 (C-21), 51.9 (C-22), 202.4 (C-23), 134.4 (C-24), 139.3 (C-25), 172.8 (C-26) and 13.9 (C-27) (See Table 1 and Supporting Information File 1). The HMBC correlations from H-24 to C-23 indicated the presence of a ketone group at C-23 (Figure 2). This conclusion was confirmed from the downfield δC of C-22 (+16.4) in comparison with 12. The relative configuration was established with the NOESY spectrum (Figure 2). Briefly, the configuration at C-5, C-9, C-10, C-13 and C-17 was determined by cross-peaks from H-28Z to H-9; H-5 to H3-19 and H3-29; H3-18 to H-9 and H-20; H3-30 to H-17; and H3-21 to H2-12. NOESY correlation between H-24 and H3-27 was not observed, suggesting that the geometry of the C-24,25 double bond was E. This was confirmed by δH comparison of H-24 with that of 1, 2, 6 and 7 (See Table 2 and Supporting Information File 1). On the basis of these spectroscopic evidences, the structure of 3 was assigned as (−)-rel-(24E)-23-oxo-3,4-seco-9βH-lanosta-4(28),7,24-triene-3,26-dioic acid.
The HRESIMS of 4, isolated as a white amorphous powder, showed a pseudomolecular [M + H]+ ion peak at m/z 471.3463, corresponding to the formula C30H46O4 (calcd. 471.3469), indicating eight degrees of insaturation. The IR absorption bands at 3416, 1708 and 1633 cm−1 suggested the presence of hydroxyl, carbonyl and olefin functionalities. The 13C NMR and DEPT-135 spectra of 4 showed signals for 30 carbons designated as five methyl; twelve methylene, including one alkene at δC 130.5; five methine, including one secondary alcohol at δC 77.1; and eight quaternary carbons, including those at δC 170.8 and 207.6 representing carboxylic and ketone carbonyls, respectively (Table 1). The 1H NMR spectrum showed two doublets at δH 0.35 (J = 3.9 Hz) and 0.52 (J = 3.9 Hz) characteristic of a cyclopropane ring (Table 2), suggesting that 4 is a member of the cycloartanes, which is an important triterpenic family in the genus Abies[3]. In the 1H–1H COSY spectrum, correlations between H2-2 to H2-1 and H-3; H2-6 to H-5 and H2-7; H2-7 to H-8; H2-16 to H2-15 and H-17; and H-20 to H3-21 and H2-22 were observed (Figure 2). HMBC correlations from H2-19 to C-1, C-5, C-8, C-9, C-10 and C-11 connected together three different fragments in the vicinity of the cyclopropyl group. Other correlations between H3-18 to C-12, C-13, C-14 and C-17; H3-21 to C-17, C-20 and C-22; H2-27 to C-24, C-25 and C-26; H3-28 and H3-29 to C-3, C-4, C-5, C-28 and C-29; H3-30 to C-8, C-13, C-14 and C-15; and H2-22 and H2-24 to C-23 were observed and completely assigned the carbon skeleton of the molecule (Figure 2). The relative configuration was determined with the help of a 2D NOESY experiment showing correlations from H-19β to H-6β, H-8 and H3-29; H-5 to H3-28 and H-6α; H3-30 to H-11α and H-17; and H-22b to H-20 and H2-16 (Figure 2). The α-orientation of the hydroxy group at C-3 was deduced from the small coupling constant of H-3 (J = 2.4 Hz), and from the NOESY correlations with both H3-28 and H3-29. Accordingly, the structure of compound 4 was defined as (+)-rel-3α-hydroxy-23-oxocycloart-25(27)-en-26-oic acid.
The absolute stereochemistry of the new compounds (1–4) has not been determined experimentally. However, the previously described compounds 7, 9, 10 and 11 have been shown to possess the usual configuration for triterpenes [8,10,11]. The structures of many other triterpenes isolated from the genus Abies were also reported with this absolute configuration according to their X-ray crystallographic data [20-22].
The structure of compound 8 was reported by Xia et al [9]. In their paper, the configuration at Δ22 was determined as E but it was not supported by any spectroscopic data. Since 1H and 13C NMR data obtained for 8 were identical to those reported by Xia within 0.01 and 0.1 ppm respectively (see Supporting Information File 1), we supposed that both molecules were the same. However, the geometry at Δ22 should be assigned to Z because of the clear NOESY correlation between H-22 and H-24. Interestingly, lanostane with E geometry at Δ22 has never been isolated so far. Moreover, triterpenes with this kind of side chain bearing an E configuration for Δ22 have only been reported by Guo et al [23,24]. During their work on Schisandra spp., they isolated many nortriterpenes having both Δ22 configurations. A statistical analysis of the 1H chemical shift for H-22 and H-24 was conducted: for E-configured Δ22, δH are 5.9 ± 0.2 and 7.8 ± 0.1 while for Z-configured Δ22, δH are 5.3 ± 0.1 and 7.2 ± 0.2, respectively. Since δH measured for compound 8 was 4.98 and 6.97, it should be assigned as (22Z)-3,4-seco-9βH-lanosta-4(28),7,22,24-tetraen-23,26-olid-3-oic acid.
The isolates were evaluated in vitro for their cytotoxic activities against two human cancer cell lines, namely lung carcinoma (A549) and colon adenocarcinoma (DLD-1), as well as against one healthy cell line (WS1) using the resazurin reduction test [25]. Etoposide was used as a positive control (IC50 ≤ 1.0 µM). None of the compounds were found to be active (IC50 > 25 µM) with the exception of abiesonic acid (6), which showed a weak cytotoxic activity against A549 (IC50 = 22 µM). The antibacterial activity of isolated compounds was also evaluated in vitro against E. coli and S. aureus using the microdilution assay [26] with gentamycin as a positive control (MIC < 0.1 µg/mL). No activity was observed for all the tested compounds (MIC ≥ 50 µM) except for triterpenoids 1 and 4, which were weakly active against S. aureus (MIC = 25 µM).
Supporting Information
Supporting Information File 1:
Experimental procedures, product characterization and 1H and 13C spectra for compounds 1–18.
We acknowledge "Chaire de Recherche sur les Agents Anticancéreux d'Origine Végétale" and NSERC for funding, and Catherine Dussault for biological assessment. The photograph from our graphical abstract is from Robert H. Mohlenbrock @ USDA-NRCS PLANTS Database / USDA NRCS. 1995. Northeast wetland flora: Field office guide to plant species. Northeast National Technical Center, Chester.
References
Mabberley, D. J. Mabberley's plant-book: a portable dictionary of plants, their classification and uses, 3rd ed.; Cambridge University Press: Cambridge, 2008; pp 1 ff.
Return to citation in text:
[1]
Takahashi, T. J. Pharm. Soc. Jpn.1938,58, 888–901.
Return to citation in text:
[1]
Yang, X.-W.; Li, S.-M.; Shen, Y.-H.; Zhang, W.-D. Chem. Biodiversity2008,5, 56–81. doi:10.1002/cbdv.200890015
Return to citation in text:
[1]
[2]
Herrick, J. W.; Snow, D. R. Iroquois medical botany; Syracuse University Press: Syracuse, NY, 1995; p 278.
Return to citation in text:
[1]
Pichette, A.; Larouche, P.-L.; Lebrun, M.; Legault, J. Phytother. Res.2006,20, 371–373. doi:10.1002/ptr.1863
Return to citation in text:
[1]
Lavoie, S.; Legault, J.; Gauthier, C.; Mshvildadze, V.; Mercier, S.; Pichette, A. Org. Lett.2012,14, 1504–1507. doi:10.1021/ol300237f
Return to citation in text:
[1]
[2]
[3]
Shang, N.; Guerrero-Analco, J. A.; Musallam, L.; Saleem, A.; Muhammad, A.; Walshe-Roussel, B.; Cuerrier, A.; Arnason, J. T.; Haddad, P. S. J. Ethnopharmacol.2012,141, 1051–1057. doi:10.1016/j.jep.2012.04.002
Return to citation in text:
[1]
Roshchin, V. I.; Raldugin, V. A.; Baranova, R. A.; Pentegova, V. A. Chem. Nat. Compd.1986,22, 613–614. doi:10.1007/BF00599284
Return to citation in text:
[1]
[2]
Wada, S.-i.; Iida, A.; Tanaka, R. J. Nat. Prod.2002,65, 1657–1659. doi:10.1021/np020282b
Return to citation in text:
[1]
[2]
[3]
[4]
Raldugin, V. A.; Gatilov, Y. V.; Rybalova, T. V.; Rashkes, Y. V. Chem. Nat. Compd.1986,22, 645–651. doi:10.1007/BF00598342
Return to citation in text:
[1]
[2]
Abad, A.; Arnó, M.; Peiró, M.; Zaragoza, R. J. Tetrahedron1991,47, 3829–3844. doi:10.1016/S0040-4020(01)80907-8
Return to citation in text:
[1]
Zhao, Y.-X.; Zhou, L.; Guo, L.; Luo, X.-D.; Zhou, J. J. Asian Nat. Prod. Res.2005,7, 259–264. doi:10.1080/10286020410001690163
Return to citation in text:
[1]
Alvarez-Manzaneda, E. J.; Chahboun, R.; Guardia, J. J.; Lachkar, M.; Dahdouh, A.; Lara, A.; Messouri, I. Tetrahedron Lett.2006,47, 2577–2580. doi:10.1016/j.tetlet.2006.02.037
Return to citation in text:
[1]
Wahlberg, I.; Vogt, C.; Wallin, I.; Nishida, T.; Enzell, C. R. Acta Chem. Scand., Ser. B1982,36, 573–576. doi:10.3891/acta.chem.scand.36b-0573
Return to citation in text:
[1]
Wahlberg, I.; Karlsson, K.; Curvall, M.; Nishida, T.; Enzell, C. R. Acta Chem. Scand., Ser. B1978,32, 203–215. doi:10.3891/acta.chem.scand.32b-0203
Return to citation in text:
[1]
Shevtsov, S. A.; Raldugin, V. A. Chem. Nat. Compd.1989,25, 182–187. doi:10.1007/BF00598407
Return to citation in text:
[1]
Li, Y.-L.; Gao, Y.-X.; Yang, X.-W.; Jin, H.-Z.; Ye, J.; Simmons, L.; Wang, N.; Steinmetz, A.; Zhang, W.-D. Phytochemistry2012,81, 159–164. doi:10.1016/j.phytochem.2012.05.032
Return to citation in text:
[1]
O'Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Eur. J. Biochem.2000,267, 5421–5426. doi:10.1046/j.1432-1327.2000.01606.x
Return to citation in text:
[1]
Banfi, E.; Scialino, G.; Monti-Bragadin, C. J. Antimicrob. Chemother.2003,52, 796–800. doi:10.1093/jac/dkg439
Return to citation in text:
[1]
Mabberley, D. J. Mabberley's plant-book: a portable dictionary of plants, their classification and uses, 3rd ed.; Cambridge University Press: Cambridge, 2008; pp 1 ff.
Shang, N.; Guerrero-Analco, J. A.; Musallam, L.; Saleem, A.; Muhammad, A.; Walshe-Roussel, B.; Cuerrier, A.; Arnason, J. T.; Haddad, P. S. J. Ethnopharmacol.2012,141, 1051–1057. doi:10.1016/j.jep.2012.04.002
Alvarez-Manzaneda, E. J.; Chahboun, R.; Guardia, J. J.; Lachkar, M.; Dahdouh, A.; Lara, A.; Messouri, I. Tetrahedron Lett.2006,47, 2577–2580. doi:10.1016/j.tetlet.2006.02.037


![[1860-5397-9-150-1]](/bjoc/content/figures/1860-5397-9-150-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-150-2]](/bjoc/content/figures/1860-5397-9-150-2.svg?scale=2.0&max-width=1024&background=FFFFFF)