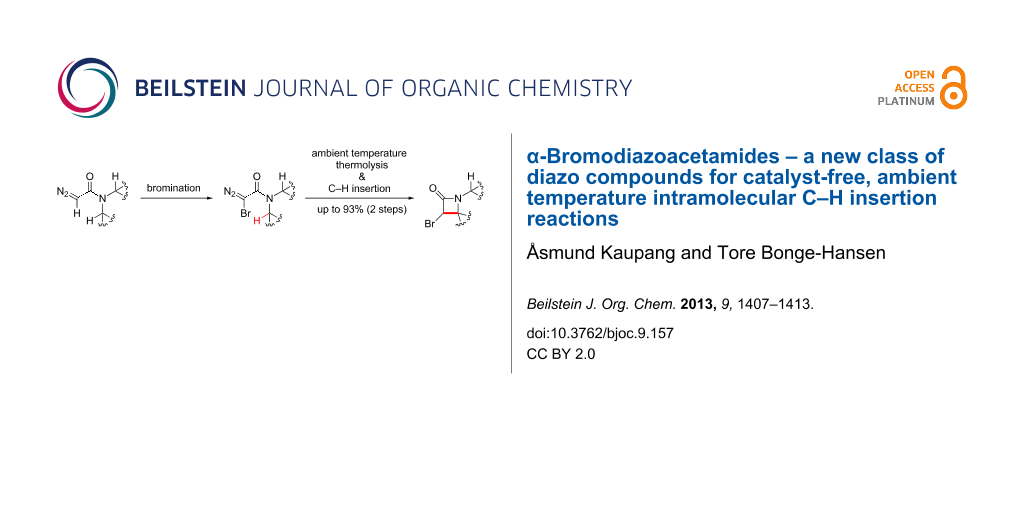
Abstract
In this work, we introduce a new class of halodiazocarbonyl compounds, α-halodiazoacetamides, which through a metal-free, ambient-temperature thermolysis perform intramolecular C–H insertions to produce α-halo-β-lactams. When carried out with α-bromodiazoacetamides bearing cyclic side chains, the thermolysis reaction affords bicyclic α-halo-β-lactams, in some cases in excellent yields, depending on the ring size and substitution pattern of the cyclic amide side chains.

Graphical Abstract
Introduction
Diazocarbonyl compounds are popular precursors for carbonylcarbenes and -carbenoids, the synthetic utility of which is thoroughly established through their successful employment in cycloaddition, ylide formation, cyclopropanation and C–H insertion reactions [1-3]. A generally useful modification of diazo compounds is the substitution of the α-hydrogen for an electrophile. This substitution can be effected in the presence of a base or starting from the metalated diazo compound, and leaves the diazo function intact [4]. Among the reported transformations are substitutions of the diazomethyl hydrogen for electrophiles based on boron [5-7], nitrogen (NO2+) [8-12], silicon [13-15], phosphorous [16-18], sulfur [19-21] and halogens [10,22-30], as well as carbon, e.g., in aldol reactions with aldehydes [31,32], ketones [33,34] and imines [35,36].
The first syntheses of α-halodiazoacetic esters, reported in the late 1960s, employed electrophilic diazoalkane substitution; the mercury or silver salts of ethyl diazoacetate (EDA) were reacted with sources of electrophilic halogen (SO2Cl2, Br2 or I2). These protocols allowed Gerhart and Schöllkopf et al. to study the properties of the resulting α-halodiazoacetates and the reactivity of their photolytically derived carbenes [26-30]. Some ten years later, Regitz et al. reported the syntheses of an α-halodiazomethyl phosphonic acid dimethyl ester and an α-halodiazomethyl diphenyl phosphoxide, starting from the silver salts of the respective diazo compounds [10].
More recently, two novel protocols for the halogenation of diazoesters and -phosphonates have been introduced by our group, both employing an N-halosuccinimide as the halogen source in combination with either the amidine base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or sodium hydride (NaH). In these reports, the obtained α-halodiazoacetates and α-halodiazophosphonates were successfully applied in dirhodium(II)-catalysed cyclopropanation, and C–H and Si–H insertion reactions [37-39].
There are, to the best of our knowledge, no reports in the literature of α-halodiazoacetamides as a substance class. Thus, we wished to expand the substrate scope of one of our published methodologies to encompass the halogenation of diazoacetamides. We report herein the bromination of the diazoacetamides derived from a selection of cyclic secondary amines, using DBU and N-bromophthalimide (NBP), as well as an investigation of the ability of the carbenes/carbenoids derived from the resulting α-bromodiazoacetamides to form α-bromo-β-lactams.
Results
The diazoacetamides 3a–f were synthesised from α-bromoacetamides 2a–f using a protocol published by Toma et al. [40], modified by exchanging the base employed in the original procedure (DBU) for 1,1,3,3-tetramethylguanidine (TMG). The use of TMG allowed for a more convenient, nonaqueous workup, involving the near quantitative removal of the produced TMG-p-toluenesulfinate salt [41] by filtration of a diethyl ether dispersion of the crude reaction mixture. This modification thus allowed for gram-scale preparations of the desired diazoacetamides. The α-bromoacetamides were in turn prepared by the acylation of the respective secondary amines 1a–f with bromoacetyl bromide (Scheme 1; see Supporting Information File 1 for full experimental details). Among the obtained diazoacetamides, 3d and 3f were not previously reported. We therefore prepared crystals and resolved their structures by single-crystal X-ray diffraction. The diazoacetamides 3d and 3f crystallised as their (Z)-rotamers. These data have recently been reported [42,43].
![[1860-5397-9-157-i1]](/bjoc/content/inline/1860-5397-9-157-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the diazoacetamides.
Scheme 1: Preparation of the diazoacetamides.
The diazoacetamides were brominated at −5 °C with NBP in the presence of DBU and passed through a dry-ice-cooled plug of silica gel with CH2Cl2 (precooled to −15 °C), in order to remove the base and phthalimide. Allowing the solution to warm to ambient temperature effected the thermolysis of the α-bromodiazoacetamides (Scheme 2). Although we have not determined the exact temperature at which the thermolysis takes place, the α-bromodiazoacetamides will rapidly lose their bright red colour at temperatures above 0 °C.
![[1860-5397-9-157-i2]](/bjoc/content/inline/1860-5397-9-157-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Bromination of the diazoacetamides 3a–f and thermolysis of the α-bromodiazoacetamides 4a–f.
Scheme 2: Bromination of the diazoacetamides 3a–f and thermolysis of the α-bromodiazoacetamides 4a–f.
As can be seen in Table 1, the obtained yields of the α-bromo-β-lactams 5a–f vary significantly throughout the series. Among the derivatives with aliphatic amide side chains, the yields increase dramatically with ring size (Table 1, entries 1–3, products 5a–c), whereas among the derivatives bearing 1,4-heterosubstituted six-membered rings as side chains, poorer yields are obtained (Table 1, entries 4–6, products 5d–f). The latter result could possibly be viewed as an expression of the deactivation of the C–H bonds β to the N-methylene groups of the amide [44]. With the exception of the piperazine derivative 5d, the observed diastereomeric ratio in the β-lactam products was approximately 6:1, favouring the diastereomer in which the bromine atom and the ring fragment are in a trans relationship (hereafter referred to as exo-5a–f). The stereochemistry of the obtained β-lactams was determined based on the previously published NMR data for endo/exo-5b [45], as well as on the characteristic magnitude of the couplings and chemical shift values of the α-protons of the endo/exo stereoisomers [46-49].
Table 1: Yieldsa of β-lactams 5a–f obtained by thermolysis of α-bromodiazoacetamides 4a–f.
| Yieldsa (%) | ||||||
|---|---|---|---|---|---|---|
| Entry | Product | exo | endo | exo + endo | exo/endo | α,α’-dibromoacetamide |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-157-i3.svg?max-width=637&scale=1.0)
5a |
7b | –c | 7b | n/a | 34 |
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-157-i4.svg?max-width=637&scale=1.0)
5b |
73 | 11 | 84 | 7:1 | 2 |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-157-i5.svg?max-width=637&scale=1.0)
5c |
77
(81)d |
16
(13)d |
93
(94)d |
5:1
(6:1)d |
trace
(–) |
| 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-157-i6.svg?max-width=637&scale=1.0)
5d |
34 | 2 | 36 | 17:1 | 21 |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-157-i7.svg?max-width=637&scale=1.0)
5e |
12 | 1 | 13 | 6:1 | 20 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-157-i8.svg?max-width=637&scale=1.0)
5f |
14 | 3 | 17 | 5:1 | 12 |
aDetermined by 1H NMR using an internal standard (see Supporting Information File 1 for details). bDecomposed in CDCl3 within 48 hours (see Supporting Information File 1 for details). cNot detected by 1H NMR. dIsolated yield after chromatography. Bromination performed with NBS/DBU.
The high-yielding C–H insertions (Table 1, entries 2–3, products 5b,c) proceed cleanly and with few byproducts (see the NMR spectra of the crude reaction mixtures containing 5b,c in Supporting Information File 1). In contrast, in the lower yielding reactions, a byproduct that could be routinely identified was the corresponding α,α'-dibromoacetamide, the origin of which remains unclear. Due to the overlap of signals from the amide side chains in the starting materials and those of eventual dimeric products, the extent of formation of carbene dimers was not possible to determine from the crude 1H NMR spectra.
As can be interpreted from Table 1, the dominant reaction pathway in the high-yielding reactions (Table 1, entries 2–3), is apparently the intramolecular C–H insertion to form a β-lactam. The preferential formation of intramolecular products from N,N’-disubstituted diazoacetamides, as compared to diazoacetates has previously been rationalised in terms of the proximity of the side chain C–H bonds to the carbene centre [50-52]. The greater ease with which the α-bromocarbene amide can insert into the C–H bonds of the larger rings (cf. the increase in yields of products 5a, 5b and 5c) suggests that the proximity and/or conformational flexibility of the C–H fragment is of importance.
The α-bromo-β-lactam 5b and its α-chloro-analogue have previously been prepared by Johansson et al. in 61% [45] and 53–54% yield [53,54], respectively, by the thermolysis of α-dihalo(phenylmercury)acetamides in bromobenzene under reflux: a reaction that plausibly occurs with a free halocarbonylcarbene or a mercury carbenoid as intermediate [45,55,56]. In the case of the α-bromo-β-lactam 5b, the authors reported an exo-diastereoselectivity of 5.25:1. In order to compare the reactivity of other halides in the α-position, we prepared and thermolysed the α-chloro- and α-iodo analogues of 4d (see results in Table 5.1 in Supporting Information File 1) [57]. In analogy to the results of Johansson et al., the α-chloro analogue afforded a lower yield of the corresponding α-chloro-β-lactam. In the case of the α-iodo analogue, the surprisingly low yield obtained may be due to decomposition during the chromatographic step, as indicated by a change in colour from red to purple, possibly owing to the formation of I2.
In a broader context, related examples of carbene/carbenoid C–H insertions to form β-lactams exist in the literature, in which the α-substituent on the carbene carbon varies (see below). The α-phenyl analogue of β-lactam 5b has previously been prepared by carbene C–H insertion. In these reports, the base-promoted decomposition of a hydrazone and subsequent thermolysis of the diazo compound in, e.g., toluene under reflux, afforded the bicyclic α-phenyl-β-lactam in up to 60% yield (6:1/exo:endo) [58-60]. Axten et al. also prepared the α-phenyl-analogues of 5a, 5c and 5e, as well as an azocane (heptamethyleneimine) derivative. The yields of the obtained β-lactams were, however, not reported [60].
Dirhodium(II) catalysis
Comparing with results obtained using dirhodium(II) catalysis, the α-H-analogue of the azepane derivative 5c was prepared in 67% yield by Doyle et al. from 3d [61]. They could also prepare the analogous azocane-derived α-H-β-lactam in 45% yield, accompanied by a 22% yield of the α-H-γ-lactam. Interestingly, the dirhodium(II)-catalysed methylene C–H insertion reactions of the smaller cyclic derivatives 3a, 3b and 3e, were unsuccessful [61]. For comparison with the thermolytic reaction, we tested the performance of a small series of dirhodium(II)-catalysts in the intramolecular C–H insertion that forms 5b. Our best result was obtained with the electron-rich dirhodium(II) carboxamidate Rh2(cap)4, affording a 44% combined yield with a 6:1 exo/endo ratio (versus 84% combined yield, 7:1 exo/endo, in the thermolysis; see Table 5.2 in Supporting Information File 1 for details). Furthermore, we observed a correlation between the electron-donating ability of the dirhodium(II) catalyst and the obtained yield of the β-lactam 5d (see Table 5.2 in Supporting Information File 1). In our hands, the more stabilised carbenoid afforded the best result, suggesting that the intramolecular C–H insertion reaction was favoured by a less reactive carbenoid. This result may provide insight into why the formation of the bicyclic system is achieved starting from the α-bromodiazoacetamide 4c, but not from the α-H diazoacetamide 3c [61] (see Discussion on the carbene-stabilising effect of halogens below) employing an electronically comparable dirhodium(II) carboxamidate catalyst (Rh2(cap)4 versus Rh2(S-MEPY)4).
Discussion
Thermolysis
To the best of our knowledge, only one previous account of the synthetic application of a thermolysis of a halodiazocarbonyl compound can be found in the literature. In this report the thermolysis of an α-bromodiazoketone was successfully employed in an intramolecular cyclopropanation reaction [62]. Historically, the thermolysis of diazocarbonyl compounds has been carried out under reflux [63-66], although examples of low temperature and ambient temperature thermolysis can be found in the case of more labile diazo compounds [14,22,23,67]. Recent examples of thermolyses of diazocarbonyl compounds, include the preparation of arylcyclopropanes (cyclopropanation) and α-arylamino esters (N–H insertion) by thermolysis of aryldiazoacetates in trifluorotoluene under reflux [68,69]. In terms of their application in synthesis, the need for prolonged heating may have narrowed the substrate scope of thermolysis reactions considerably, contributing to the limited number of reports of catalyst-free thermolyses of diazo compounds in the literature. In comparison, the capability of the α-bromodiazoacetamides to thermolyse at ambient temperature without the use of a metal catalyst, offers the reactivity of the bromocarbene amides under genuinely mild reaction conditions.
Halocarbonyl carbenes and carbene/carbenoid stabilisation
In their ground state, the halocarbonyl carbenes derived from halodiazoamides are, in analogy with those derived from halodiazoesters, predicted by theory to be singlet carbenes [70]. Furthermore, in analogy to α-bromoethoxycarbonyl carbene [28,30,71], they should be electrophilic carbenes. An important modulator of carbene electrophilicity is the stabilising effect of π-donation from a substituent on the carbene carbon into the vacant carbene 2p-orbital. This effect is predicted by theory, as well as observed experimentally [28,30,71-73]. The degree of stabilisation exerted by a bromine substituent has been quantified in the scale of carbenic reactivity, introduced by R. A. Moss [71]. The increased stabilisation of α-bromocarboethoxycarbene as compared to α-H-carboethoxycarbene, is apparent in its increased propensity towards reaction with the more electron-rich of the available reaction partners, a propensity which has traditionally been quantified by the ratios of cyclopropanation of increasingly substituted ethylenes [28,30,71]. More recently, a two-dimensional carbenic reactivity surface has been established by Brinker and co-workers. Unfortunately, no halocarbonylcarbene has yet been included in these studies [73].
The donation of electron density to the vacant carbenoid 2p orbital, has also been a topic in the paradigm of dirhodium(II) catalysis, in which the electron-donating ability of the catalyst ligands correlates to the observed selectivity of the dirhodium(II) carbenoid. Thus, in analogy to the preference for more highly substituted ethylenes, in a case where only C–H insertion is viable, a highly stabilised dirhodium(II) carbenoid shows increased selectivity among the possible C–H insertion partners. For alkanes, the following order of preference is observed: R3CH > R2CH2 > RCH3 [2,74-76]. The order of reactivity of the C–H bonds reflects the varying abilities of the CHn carbons to stabilise the build-up of positive charge as the incipient electrophilic carbene/carbenoid 2p orbital attacks the C–H σ-bonding orbital of the substrate. Consequently, C–H bonds vicinal to heteroatoms are activated for insertion [44,77]. The observed selectivity of the more stabilised carbenoids is attributed to the donation of electron density from the 4d orbitals of the dirhodium(II) complex to the vacant carbenoid 2p orbital [76,78-81], which attenuates its electrophilicity. This effect is analogous to the increased selectivity conferred by π-donation from a halogen substituent on the carbene carbon (see above) [28,30,71,81-84].
Future perspectives
A pronounced goal in the previously published syntheses of the piperidine-derived bicyclic β-lactams, analogues of 5b, was to develop a model system for the synthesis of β-lactam antibiotics [58,85]. In this vein, the metal-independent character of the presented thermolysis of α-bromodiazoacetamides makes the transformation compatible with late-stage synthesis in medicinal chemistry. Additionally, in a preparative context, the formation of α-bromo-β-lactams such as 5b,c using thermolysis is advantageous; 5c could be conveniently prepared in excellent isolated yields from 3c, without the use of expensive catalysts, inert atmosphere or anhydrous conditions.
Conclusion
We have demonstrated the halogenation of a series of diazoacetamides and the ambient temperature thermolysis of the resulting α-halodiazoacetamides. The synthetic utility of the α-bromodiazoacetamides has been demonstrated in the preparation of bicyclic α-bromo-β-lactams. In the cases where C–H insertion is conformationally favoured, the α-bromo-β-lactams were obtained in good to excellent yields.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and physical data for the obtained products. | ||
| Format: PDF | Size: 4.9 MB | Download |
Acknowledgements
The authors wish to thank senior engineer Dr. Osamu Sekiguchi for the recording of mass-spectrometry data, Professor Carl-Henrik Gørbitz for assistance with the X-ray crystallography, and Dr. Tor Erik Kristensen, Dr. Christian Schnaars and Dr. Martin Hennum for their competent input and advice throughout the preparation of this manuscript.
References
-
Ye, T.; McKervey, M. A. Chem. Rev. 1994, 94, 1091–1160. doi:10.1021/cr00028a010
Return to citation in text: [1] -
Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; John Wiley & Sons: New York, NY, 1998.
Return to citation in text: [1] [2] -
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n
Return to citation in text: [1] -
Fink, J.; Regitz, M. Synthesis 1985, 569–585. doi:10.1055/s-1985-34147
Return to citation in text: [1] -
Arthur, M. P.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 1991, 113, 5856–5857. doi:10.1021/ja00015a046
Return to citation in text: [1] -
Ansorge, A.; Brauer, D. J.; Bürger, H.; Hagen, T.; Pawelke, G. Angew. Chem., Int. Ed. Engl. 1993, 32, 384–385. doi:10.1002/anie.199303841
Angew. Chem. 1993, 105, 429–430. doi:10.1002/ange.19931050319
Return to citation in text: [1] -
Weber, L.; Wartig, H. B.; Stammler, H.-G.; Neumann, B. Organometallics 2001, 20, 5248–5250. doi:10.1021/om010588y
Return to citation in text: [1] -
Schöllkopf, U.; Schäfer, H. Angew. Chem., Int. Ed. Engl. 1965, 4, 358. doi:10.1002/anie.196503581
Angew. Chem. 1965, 77, 379. doi:10.1002/ange.19650770808
Return to citation in text: [1] -
Schöllkopf, U.; Tonne, P.; Schäfer, H.; Markusch, P. Justus Liebigs Ann. Chem. 1969, 722, 45–51. doi:10.1002/jlac.19697220107
Return to citation in text: [1] -
Regitz, M.; Weber, B.; Eckstein, U. Liebigs Ann. Chem. 1979, 1002–1019. doi:10.1002/jlac.197919790711
Return to citation in text: [1] [2] [3] -
O'Bannon, P. E.; Dailey, W. P. Tetrahedron Lett. 1989, 30, 4197–4200. doi:10.1016/S0040-4039(01)80688-2
Return to citation in text: [1] -
Ivanova, O. A.; Yashin, N. V.; Averina, E. B.; Grishin, Yu. K.; Kuznetsova, T. S.; Zefirov, N. S. Russ. Chem. Bull. 2001, 50, 2101–2105. doi:10.1023/A:1015045216923
Return to citation in text: [1] -
Schöllkopf, U.; Frasnelli, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 301–302. doi:10.1002/anie.197003011
Angew. Chem. 1970, 82, 291. doi:10.1002/ange.19700820705
Return to citation in text: [1] -
Maas, G.; Brueckmann, R. J. Org. Chem. 1985, 50, 2801–2802. doi:10.1021/jo00215a049
Return to citation in text: [1] [2] -
Austin, W. F.; Zhang, Y.; Danheiser, R. L. Tetrahedron 2008, 64, 915–925. doi:10.1016/j.tet.2007.10.113
Return to citation in text: [1] -
Baceiredo, A.; Bertrand, G.; Sicard, G. J. Am. Chem. Soc. 1985, 107, 4781–4783. doi:10.1021/ja00302a032
Return to citation in text: [1] -
Nees, H.-J.; Keller, H.; Facklam, T.; Herrmann, A.; Welsch, J.; Bergsträßer, U.; Heydt, H.; Regitz, M. J. Prakt. Chem. 1993, 335, 589–598. doi:10.1002/prac.19933350704
Return to citation in text: [1] -
Krysiak, J.; Lyon, C.; Baceiredo, A.; Gornitzka, H.; Mikolajczyk, M.; Bertrand, G. Chem.–Eur. J. 2004, 10, 1982–1986. doi:10.1002/chem.200305722
Return to citation in text: [1] -
Weiss, R.; Handke, M.; Reichel, S.; Hampel, F. Z. Naturforsch., B: Chem. Sci. 1998, 53b, 599–619.
Return to citation in text: [1] -
Cuevas-Yañez, E.; Muchowski, J. M.; Cruz-Almanza, R. Tetrahedron Lett. 2004, 45, 2417–2419. doi:10.1016/j.tetlet.2004.01.074
Return to citation in text: [1] -
Wagner, T.; Lange, J.; Grote, D.; Sander, W.; Schaumann, E.; Adiwidjaja, G.; Adam, A.; Kopf, J. Eur. J. Org. Chem. 2009, 5198–5207. doi:10.1002/ejoc.200900482
Return to citation in text: [1] -
Closs, G. L.; Coyle, J. J. J. Am. Chem. Soc. 1962, 84, 4350. doi:10.1021/ja00881a034
Return to citation in text: [1] [2] -
Closs, G. L.; Coyle, J. J. J. Am. Chem. Soc. 1965, 87, 4270–4279. doi:10.1021/ja00947a010
Return to citation in text: [1] [2] -
Bussey, R. J.; Neuman, R. C., Jr. J. Org. Chem. 1969, 34, 1323–1327. doi:10.1021/jo01257a026
Return to citation in text: [1] -
Fridman, A.; Kolobov, N.; Zalesov, V.; Sivkova, M. Zh. Org. Khim. 1974, 10, 884–885.
Return to citation in text: [1] -
Gerhart, F.; Schöllkopf, U.; Schumacher, H. Angew. Chem., Int. Ed. Engl. 1967, 6, 74–75. doi:10.1002/anie.196700742
Angew. Chem. 1967, 79, 50. doi:10.1002/ange.19670790106
Return to citation in text: [1] [2] -
Schöllkopf, U.; Gerhart, F.; Reetz, M.; Frasnelli, H.; Schumacher, H. Justus Liebigs Ann. Chem. 1968, 716, 204–206. doi:10.1002/jlac.19687160129
Return to citation in text: [1] [2] -
Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Schöllkopf, U.; Rieber, N. Chem. Ber. 1969, 102, 488–493. doi:10.1002/cber.19691020216
Return to citation in text: [1] [2] -
Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Xiao, F.; Liu, Y.; Wang, J. Tetrahedron Lett. 2007, 48, 1147–1149. doi:10.1016/j.tetlet.2006.12.062
Return to citation in text: [1] -
Trost, B. M.; Malhotra, S.; Fried, B. A. J. Am. Chem. Soc. 2009, 131, 1674–1675. doi:10.1021/ja809181m
Return to citation in text: [1] -
Schöllkopf, U.; Bánhidai, B.; Frasnelli, H.; Meyer, R.; Beckhaus, H. Justus Liebigs Ann. Chem. 1974, 11, 1767–1783. doi:10.1002/jlac.197419741106
Return to citation in text: [1] -
Benfatti, F.; Yilmaz, S.; Cozzi, P. G. Adv. Synth. Catal. 2009, 351, 1763–1767. doi:10.1002/adsc.200900435
Return to citation in text: [1] -
Jiang, N.; Wang, J. Tetrahedron Lett. 2002, 43, 1285–1287. doi:10.1016/S0040-4039(01)02375-9
Return to citation in text: [1] -
Kantam, M. L.; Balasubrahmanyam, V.; Kumar, K. B. S.; Venkanna, G. T.; Figueras, F. Adv. Synth. Catal. 2007, 349, 1887–1890. doi:10.1002/adsc.200600597
Return to citation in text: [1] -
Bonge, H. T.; Pintea, B.; Hansen, T. Org. Biomol. Chem. 2008, 6, 3670–3672. doi:10.1039/b814374a
Return to citation in text: [1] -
Bonge, H. T.; Hansen, T. Synthesis 2009, 91–96. doi:10.1055/s-0028-1083272
Return to citation in text: [1] -
Schnaars, C.; Hansen, T. Org. Lett. 2012, 14, 2794–2797. doi:10.1021/ol3010276
Return to citation in text: [1] -
Toma, T.; Shimokawa, J.; Fukuyama, T. Org. Lett. 2007, 9, 3195–3197. doi:10.1021/ol701432k
Return to citation in text: [1] -
Kaupang, Å.; Görbitz, C. H.; Bonge-Hansen, T. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2013, 69, in press. doi:10.1107/S0108270113015011
Return to citation in text: [1] -
Kaupang, Å.; Görbitz, C. H.; Hansen, T. Acta Crystallogr., Sect. E: Struct. Rep. Online 2010, 66, o1299. doi:10.1107/S1600536810016211
Return to citation in text: [1] -
Kaupang, Å.; Görbitz, C. H.; Hansen, T. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o1844–o1845. doi:10.1107/S1600536811023774
Return to citation in text: [1] -
Davies, H. M. L.; Yang, J. Adv. Synth. Catal. 2003, 345, 1133–1138. doi:10.1002/adsc.200303089
Return to citation in text: [1] [2] -
Johansson, N. G.; Åkermark, B. Acta Chem. Scand. 1971, 25, 1927–1929. doi:10.3891/acta.chem.scand.25-1927
Return to citation in text: [1] [2] [3] -
Kagan, H. B.; Basselier, J.-J.; Luche, J.-L. Tetrahedron Lett. 1964, 5, 941–948. doi:10.1016/S0040-4039(00)90411-8
Return to citation in text: [1] -
Barrow, K. D.; Spotswood, T. M. Tetrahedron Lett. 1965, 6, 3325–3335. doi:10.1016/S0040-4039(01)89203-0
Return to citation in text: [1] -
McMillan, I.; Stoodley, R. J. Tetrahedron Lett. 1966, 7, 1205–1210. doi:10.1016/S0040-4039(00)72394-X
Return to citation in text: [1] -
Decazes, J.; Luche, J. L.; Kagan, H. B. Tetrahedron Lett. 1970, 11, 3661–3664. doi:10.1016/S0040-4039(01)98555-7
Return to citation in text: [1] -
Rando, R. R. J. Am. Chem. Soc. 1970, 92, 6706–6707. doi:10.1021/ja00725a091
Return to citation in text: [1] -
Rando, R. R. J. Am. Chem. Soc. 1972, 94, 1629–1631. doi:10.1021/ja00760a033
Return to citation in text: [1] -
Tomioka, H.; Kondo, M.; Izawa, Y. J. Org. Chem. 1981, 46, 1090–1094. doi:10.1021/jo00319a010
Return to citation in text: [1] -
Johansson, N. G. Acta Chem. Scand. 1973, 27, 1417–1420. doi:10.3891/acta.chem.scand.27-1417
Return to citation in text: [1] -
Åkermark, B.; Byström, S.; Florin, E.; Johansson, N.-G.; Lagerlund, I. Acta Chem. Scand., Ser. B 1974, 28, 375–376. doi:10.3891/acta.chem.scand.28b-0375
Return to citation in text: [1] -
Seyferth, D.; Burlitch, J. M.; Dertouzos, H.; Simmons, H. D., Jr. J. Organomet. Chem. 1967, 7, 405–413. doi:10.1016/S0022-328X(00)85362-0
Return to citation in text: [1] -
Seyferth, D. Acc. Chem. Res. 1972, 5, 65–74. doi:10.1021/ar50050a004
Return to citation in text: [1] -
Kaupang, Å., Intramolecular C–H insertion reactions of α-bromodiazoacetamides. M.Sc. Thesis, University of Oslo, Norway, 2010. http://hdl.handle.net/10852/12702
Return to citation in text: [1] -
Corey, E. J.; Felix, A. M. J. Am. Chem. Soc. 1965, 87, 2518–2519. doi:10.1021/ja01089a055
Return to citation in text: [1] [2] -
Earle, R. H.; Hurst, D. T.; Viney, M. J. Chem. Soc. C 1969, 2093–2098. doi:10.1039/J39690002093
Return to citation in text: [1] -
Axten, J. M.; Krim, L.; Kung, H. F.; Winkler, J. D. J. Org. Chem. 1998, 63, 9628–9629. doi:10.1021/jo982214t
Return to citation in text: [1] [2] -
Doyle, M. P.; Kalinin, A. V. Synlett 1995, 1075–1076. doi:10.1055/s-1995-5176
Return to citation in text: [1] [2] [3] -
Balci, M.; Schalenbach, R.; Vogel, E. Angew. Chem., Int. Ed. Engl. 1981, 20, 809–811. doi:10.1002/anie.198108091
Angew. Chem. 1981, 93, 816–818. doi:10.1002/ange.19810930932
Return to citation in text: [1] -
Dave, V.; Warnhoff, E. W. Org. React. 1970, 18, 217–401. doi:10.1002/0471264180.or018.03
Return to citation in text: [1] -
Burke, S. D.; Grieco, P. A. Org. React. 1979, 26, 361–475. doi:10.1002/0471264180.or026.02
Return to citation in text: [1] -
Schöllkopf, U.; Wiskott, E. Justus Liebigs Ann. Chem. 1966, 694, 44–55. doi:10.1002/jlac.19666940107
Return to citation in text: [1] -
Schöllkopf, U.; Tonne, P. Justus Liebigs Ann. Chem. 1971, 753, 135–142. doi:10.1002/jlac.19717530112
Return to citation in text: [1] -
Schöllkopf, U.; Markusch, P. Justus Liebigs Ann. Chem. 1971, 753, 143–150. doi:10.1002/jlac.19717530113
Return to citation in text: [1] -
Ovalles, S. R.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2011, 13, 4284–4287. doi:10.1021/ol201628d
Return to citation in text: [1] -
Hansen, S. R.; Spangler, J. E.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2012, 14, 4626–4629. doi:10.1021/ol3020754
Return to citation in text: [1] -
Tippmann, E. M. Studies of carbene-solvent interactions. Ph.D. Thesis, Ohio State University, Columbus Ohio, U.S.A., 2003.
http://hdl.handle.net/2374.OX/5847
Return to citation in text: [1] -
Moss, R. A.; Mallon, C. B.; Ho, C.-T. J. Am. Chem. Soc. 1977, 99, 4105–4110. doi:10.1021/ja00454a032
Return to citation in text: [1] [2] [3] [4] [5] -
Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39–92. doi:10.1021/cr940472u
Return to citation in text: [1] -
Mieusset, J.-L.; Brinker, U. H. J. Org. Chem. 2008, 73, 1553–1558. doi:10.1021/jo7026118
Return to citation in text: [1] [2] -
Doyle, M. P.; Westrum, L. J.; Wolthuis, W. N. E.; See, M. M.; Boone, W. P.; Bagheri, V.; Pearson, M. M. J. Am. Chem. Soc. 1993, 115, 958–964. doi:10.1021/ja00056a021
Return to citation in text: [1] -
Padwa, A.; Austin, D. J.; Price, A. T.; Semones, M. A.; Doyle, M. P.; Protopopova, M. N.; Winchester, W. R.; Tran, A. J. Am. Chem. Soc. 1993, 115, 8669–8680. doi:10.1021/ja00072a021
Return to citation in text: [1] -
Pirrung, M. C.; Morehead, A. T., Jr. J. Am. Chem. Soc. 1994, 116, 8991–9000. doi:10.1021/ja00099a017
Return to citation in text: [1] [2] -
Davies, H. M. L.; Venkataramani, C.; Hansen, T.; Hopper, D. W. J. Am. Chem. Soc. 2003, 125, 6462–6468. doi:10.1021/ja0290072
Return to citation in text: [1] -
Doyle, M. P.; Ren, T. The Influence of Ligands on Dirhodium(II) on Reactivity and Selectivity in Metal Carbene Reactions. In Progress in Inorganic Chemistry; Karlin, K. D., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001; pp 113–168.
Return to citation in text: [1] -
Lloret, J.; Carbo, J. J.; Bo, C.; Lledos, A.; Perez-Prieto, J. Organometallics 2008, 27, 2873–2876. doi:10.1021/om800089k
Return to citation in text: [1] -
Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o
Return to citation in text: [1] -
Bonge, H. T.; Hansen, T. J. Org. Chem. 2010, 75, 2309–2320. doi:10.1021/jo100113b
Return to citation in text: [1] [2] -
Bonge, H. T.; Hansen, T. Eur. J. Org. Chem. 2010, 4355–4359. doi:10.1002/ejoc.201000531
Return to citation in text: [1] -
Bonge, H. T.; Hansen, T. Tetrahedron Lett. 2010, 51, 5378–5381. doi:10.1016/j.tetlet.2010.07.116
Return to citation in text: [1] -
Bonge, H. T.; Hansen, T. Pure Appl. Chem. 2011, 83, 565–575. doi:10.1351/PAC-CON-10-10-18
Return to citation in text: [1] -
Johansson, N. G.; Åkermark, B. Tetrahedron Lett. 1971, 12, 4785–4786. doi:10.1016/S0040-4039(01)97615-4
Return to citation in text: [1]
| 61. | Doyle, M. P.; Kalinin, A. V. Synlett 1995, 1075–1076. doi:10.1055/s-1995-5176 |
| 61. | Doyle, M. P.; Kalinin, A. V. Synlett 1995, 1075–1076. doi:10.1055/s-1995-5176 |
| 61. | Doyle, M. P.; Kalinin, A. V. Synlett 1995, 1075–1076. doi:10.1055/s-1995-5176 |
| 1. | Ye, T.; McKervey, M. A. Chem. Rev. 1994, 94, 1091–1160. doi:10.1021/cr00028a010 |
| 2. | Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; John Wiley & Sons: New York, NY, 1998. |
| 3. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 13. |
Schöllkopf, U.; Frasnelli, H. Angew. Chem., Int. Ed. Engl. 1970, 9, 301–302. doi:10.1002/anie.197003011
Angew. Chem. 1970, 82, 291. doi:10.1002/ange.19700820705 |
| 14. | Maas, G.; Brueckmann, R. J. Org. Chem. 1985, 50, 2801–2802. doi:10.1021/jo00215a049 |
| 15. | Austin, W. F.; Zhang, Y.; Danheiser, R. L. Tetrahedron 2008, 64, 915–925. doi:10.1016/j.tet.2007.10.113 |
| 40. | Toma, T.; Shimokawa, J.; Fukuyama, T. Org. Lett. 2007, 9, 3195–3197. doi:10.1021/ol701432k |
| 28. | Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9 |
| 30. | Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409 |
| 71. | Moss, R. A.; Mallon, C. B.; Ho, C.-T. J. Am. Chem. Soc. 1977, 99, 4105–4110. doi:10.1021/ja00454a032 |
| 72. | Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39–92. doi:10.1021/cr940472u |
| 73. | Mieusset, J.-L.; Brinker, U. H. J. Org. Chem. 2008, 73, 1553–1558. doi:10.1021/jo7026118 |
| 8. |
Schöllkopf, U.; Schäfer, H. Angew. Chem., Int. Ed. Engl. 1965, 4, 358. doi:10.1002/anie.196503581
Angew. Chem. 1965, 77, 379. doi:10.1002/ange.19650770808 |
| 9. | Schöllkopf, U.; Tonne, P.; Schäfer, H.; Markusch, P. Justus Liebigs Ann. Chem. 1969, 722, 45–51. doi:10.1002/jlac.19697220107 |
| 10. | Regitz, M.; Weber, B.; Eckstein, U. Liebigs Ann. Chem. 1979, 1002–1019. doi:10.1002/jlac.197919790711 |
| 11. | O'Bannon, P. E.; Dailey, W. P. Tetrahedron Lett. 1989, 30, 4197–4200. doi:10.1016/S0040-4039(01)80688-2 |
| 12. | Ivanova, O. A.; Yashin, N. V.; Averina, E. B.; Grishin, Yu. K.; Kuznetsova, T. S.; Zefirov, N. S. Russ. Chem. Bull. 2001, 50, 2101–2105. doi:10.1023/A:1015045216923 |
| 41. | Kaupang, Å.; Görbitz, C. H.; Bonge-Hansen, T. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2013, 69, in press. doi:10.1107/S0108270113015011 |
| 71. | Moss, R. A.; Mallon, C. B.; Ho, C.-T. J. Am. Chem. Soc. 1977, 99, 4105–4110. doi:10.1021/ja00454a032 |
| 5. | Arthur, M. P.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 1991, 113, 5856–5857. doi:10.1021/ja00015a046 |
| 6. |
Ansorge, A.; Brauer, D. J.; Bürger, H.; Hagen, T.; Pawelke, G. Angew. Chem., Int. Ed. Engl. 1993, 32, 384–385. doi:10.1002/anie.199303841
Angew. Chem. 1993, 105, 429–430. doi:10.1002/ange.19931050319 |
| 7. | Weber, L.; Wartig, H. B.; Stammler, H.-G.; Neumann, B. Organometallics 2001, 20, 5248–5250. doi:10.1021/om010588y |
| 10. | Regitz, M.; Weber, B.; Eckstein, U. Liebigs Ann. Chem. 1979, 1002–1019. doi:10.1002/jlac.197919790711 |
| 70. |
Tippmann, E. M. Studies of carbene-solvent interactions. Ph.D. Thesis, Ohio State University, Columbus Ohio, U.S.A., 2003.
http://hdl.handle.net/2374.OX/5847 |
| 37. | Bonge, H. T.; Pintea, B.; Hansen, T. Org. Biomol. Chem. 2008, 6, 3670–3672. doi:10.1039/b814374a |
| 38. | Bonge, H. T.; Hansen, T. Synthesis 2009, 91–96. doi:10.1055/s-0028-1083272 |
| 39. | Schnaars, C.; Hansen, T. Org. Lett. 2012, 14, 2794–2797. doi:10.1021/ol3010276 |
| 28. | Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9 |
| 30. | Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409 |
| 71. | Moss, R. A.; Mallon, C. B.; Ho, C.-T. J. Am. Chem. Soc. 1977, 99, 4105–4110. doi:10.1021/ja00454a032 |
| 31. | Xiao, F.; Liu, Y.; Wang, J. Tetrahedron Lett. 2007, 48, 1147–1149. doi:10.1016/j.tetlet.2006.12.062 |
| 32. | Trost, B. M.; Malhotra, S.; Fried, B. A. J. Am. Chem. Soc. 2009, 131, 1674–1675. doi:10.1021/ja809181m |
| 35. | Jiang, N.; Wang, J. Tetrahedron Lett. 2002, 43, 1285–1287. doi:10.1016/S0040-4039(01)02375-9 |
| 36. | Kantam, M. L.; Balasubrahmanyam, V.; Kumar, K. B. S.; Venkanna, G. T.; Figueras, F. Adv. Synth. Catal. 2007, 349, 1887–1890. doi:10.1002/adsc.200600597 |
| 14. | Maas, G.; Brueckmann, R. J. Org. Chem. 1985, 50, 2801–2802. doi:10.1021/jo00215a049 |
| 22. | Closs, G. L.; Coyle, J. J. J. Am. Chem. Soc. 1962, 84, 4350. doi:10.1021/ja00881a034 |
| 23. | Closs, G. L.; Coyle, J. J. J. Am. Chem. Soc. 1965, 87, 4270–4279. doi:10.1021/ja00947a010 |
| 67. | Schöllkopf, U.; Markusch, P. Justus Liebigs Ann. Chem. 1971, 753, 143–150. doi:10.1002/jlac.19717530113 |
| 10. | Regitz, M.; Weber, B.; Eckstein, U. Liebigs Ann. Chem. 1979, 1002–1019. doi:10.1002/jlac.197919790711 |
| 22. | Closs, G. L.; Coyle, J. J. J. Am. Chem. Soc. 1962, 84, 4350. doi:10.1021/ja00881a034 |
| 23. | Closs, G. L.; Coyle, J. J. J. Am. Chem. Soc. 1965, 87, 4270–4279. doi:10.1021/ja00947a010 |
| 24. | Bussey, R. J.; Neuman, R. C., Jr. J. Org. Chem. 1969, 34, 1323–1327. doi:10.1021/jo01257a026 |
| 25. | Fridman, A.; Kolobov, N.; Zalesov, V.; Sivkova, M. Zh. Org. Khim. 1974, 10, 884–885. |
| 26. |
Gerhart, F.; Schöllkopf, U.; Schumacher, H. Angew. Chem., Int. Ed. Engl. 1967, 6, 74–75. doi:10.1002/anie.196700742
Angew. Chem. 1967, 79, 50. doi:10.1002/ange.19670790106 |
| 27. | Schöllkopf, U.; Gerhart, F.; Reetz, M.; Frasnelli, H.; Schumacher, H. Justus Liebigs Ann. Chem. 1968, 716, 204–206. doi:10.1002/jlac.19687160129 |
| 28. | Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9 |
| 29. | Schöllkopf, U.; Rieber, N. Chem. Ber. 1969, 102, 488–493. doi:10.1002/cber.19691020216 |
| 30. | Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409 |
| 26. |
Gerhart, F.; Schöllkopf, U.; Schumacher, H. Angew. Chem., Int. Ed. Engl. 1967, 6, 74–75. doi:10.1002/anie.196700742
Angew. Chem. 1967, 79, 50. doi:10.1002/ange.19670790106 |
| 27. | Schöllkopf, U.; Gerhart, F.; Reetz, M.; Frasnelli, H.; Schumacher, H. Justus Liebigs Ann. Chem. 1968, 716, 204–206. doi:10.1002/jlac.19687160129 |
| 28. | Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9 |
| 29. | Schöllkopf, U.; Rieber, N. Chem. Ber. 1969, 102, 488–493. doi:10.1002/cber.19691020216 |
| 30. | Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409 |
| 68. | Ovalles, S. R.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2011, 13, 4284–4287. doi:10.1021/ol201628d |
| 69. | Hansen, S. R.; Spangler, J. E.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2012, 14, 4626–4629. doi:10.1021/ol3020754 |
| 19. | Weiss, R.; Handke, M.; Reichel, S.; Hampel, F. Z. Naturforsch., B: Chem. Sci. 1998, 53b, 599–619. |
| 20. | Cuevas-Yañez, E.; Muchowski, J. M.; Cruz-Almanza, R. Tetrahedron Lett. 2004, 45, 2417–2419. doi:10.1016/j.tetlet.2004.01.074 |
| 21. | Wagner, T.; Lange, J.; Grote, D.; Sander, W.; Schaumann, E.; Adiwidjaja, G.; Adam, A.; Kopf, J. Eur. J. Org. Chem. 2009, 5198–5207. doi:10.1002/ejoc.200900482 |
| 62. |
Balci, M.; Schalenbach, R.; Vogel, E. Angew. Chem., Int. Ed. Engl. 1981, 20, 809–811. doi:10.1002/anie.198108091
Angew. Chem. 1981, 93, 816–818. doi:10.1002/ange.19810930932 |
| 16. | Baceiredo, A.; Bertrand, G.; Sicard, G. J. Am. Chem. Soc. 1985, 107, 4781–4783. doi:10.1021/ja00302a032 |
| 17. | Nees, H.-J.; Keller, H.; Facklam, T.; Herrmann, A.; Welsch, J.; Bergsträßer, U.; Heydt, H.; Regitz, M. J. Prakt. Chem. 1993, 335, 589–598. doi:10.1002/prac.19933350704 |
| 18. | Krysiak, J.; Lyon, C.; Baceiredo, A.; Gornitzka, H.; Mikolajczyk, M.; Bertrand, G. Chem.–Eur. J. 2004, 10, 1982–1986. doi:10.1002/chem.200305722 |
| 33. | Schöllkopf, U.; Bánhidai, B.; Frasnelli, H.; Meyer, R.; Beckhaus, H. Justus Liebigs Ann. Chem. 1974, 11, 1767–1783. doi:10.1002/jlac.197419741106 |
| 34. | Benfatti, F.; Yilmaz, S.; Cozzi, P. G. Adv. Synth. Catal. 2009, 351, 1763–1767. doi:10.1002/adsc.200900435 |
| 63. | Dave, V.; Warnhoff, E. W. Org. React. 1970, 18, 217–401. doi:10.1002/0471264180.or018.03 |
| 64. | Burke, S. D.; Grieco, P. A. Org. React. 1979, 26, 361–475. doi:10.1002/0471264180.or026.02 |
| 65. | Schöllkopf, U.; Wiskott, E. Justus Liebigs Ann. Chem. 1966, 694, 44–55. doi:10.1002/jlac.19666940107 |
| 66. | Schöllkopf, U.; Tonne, P. Justus Liebigs Ann. Chem. 1971, 753, 135–142. doi:10.1002/jlac.19717530112 |
| 45. | Johansson, N. G.; Åkermark, B. Acta Chem. Scand. 1971, 25, 1927–1929. doi:10.3891/acta.chem.scand.25-1927 |
| 42. | Kaupang, Å.; Görbitz, C. H.; Hansen, T. Acta Crystallogr., Sect. E: Struct. Rep. Online 2010, 66, o1299. doi:10.1107/S1600536810016211 |
| 43. | Kaupang, Å.; Görbitz, C. H.; Hansen, T. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o1844–o1845. doi:10.1107/S1600536811023774 |
| 28. | Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9 |
| 30. | Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409 |
| 71. | Moss, R. A.; Mallon, C. B.; Ho, C.-T. J. Am. Chem. Soc. 1977, 99, 4105–4110. doi:10.1021/ja00454a032 |
| 44. | Davies, H. M. L.; Yang, J. Adv. Synth. Catal. 2003, 345, 1133–1138. doi:10.1002/adsc.200303089 |
| 73. | Mieusset, J.-L.; Brinker, U. H. J. Org. Chem. 2008, 73, 1553–1558. doi:10.1021/jo7026118 |
| 2. | Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; John Wiley & Sons: New York, NY, 1998. |
| 74. | Doyle, M. P.; Westrum, L. J.; Wolthuis, W. N. E.; See, M. M.; Boone, W. P.; Bagheri, V.; Pearson, M. M. J. Am. Chem. Soc. 1993, 115, 958–964. doi:10.1021/ja00056a021 |
| 75. | Padwa, A.; Austin, D. J.; Price, A. T.; Semones, M. A.; Doyle, M. P.; Protopopova, M. N.; Winchester, W. R.; Tran, A. J. Am. Chem. Soc. 1993, 115, 8669–8680. doi:10.1021/ja00072a021 |
| 76. | Pirrung, M. C.; Morehead, A. T., Jr. J. Am. Chem. Soc. 1994, 116, 8991–9000. doi:10.1021/ja00099a017 |
| 58. | Corey, E. J.; Felix, A. M. J. Am. Chem. Soc. 1965, 87, 2518–2519. doi:10.1021/ja01089a055 |
| 59. | Earle, R. H.; Hurst, D. T.; Viney, M. J. Chem. Soc. C 1969, 2093–2098. doi:10.1039/J39690002093 |
| 60. | Axten, J. M.; Krim, L.; Kung, H. F.; Winkler, J. D. J. Org. Chem. 1998, 63, 9628–9629. doi:10.1021/jo982214t |
| 60. | Axten, J. M.; Krim, L.; Kung, H. F.; Winkler, J. D. J. Org. Chem. 1998, 63, 9628–9629. doi:10.1021/jo982214t |
| 45. | Johansson, N. G.; Åkermark, B. Acta Chem. Scand. 1971, 25, 1927–1929. doi:10.3891/acta.chem.scand.25-1927 |
| 55. | Seyferth, D.; Burlitch, J. M.; Dertouzos, H.; Simmons, H. D., Jr. J. Organomet. Chem. 1967, 7, 405–413. doi:10.1016/S0022-328X(00)85362-0 |
| 56. | Seyferth, D. Acc. Chem. Res. 1972, 5, 65–74. doi:10.1021/ar50050a004 |
| 57. | Kaupang, Å., Intramolecular C–H insertion reactions of α-bromodiazoacetamides. M.Sc. Thesis, University of Oslo, Norway, 2010. http://hdl.handle.net/10852/12702 |
| 45. | Johansson, N. G.; Åkermark, B. Acta Chem. Scand. 1971, 25, 1927–1929. doi:10.3891/acta.chem.scand.25-1927 |
| 28. | Schöllkopf, U.; Reetz, M. Tetrahedron Lett. 1969, 10, 1541–1544. doi:10.1016/S0040-4039(01)87939-9 |
| 30. | Reetz, M.; Schöllkopf, U.; Bánhidai, B. Justus Liebigs Ann. Chem. 1973, 599–610. doi:10.1002/jlac.197319730409 |
| 71. | Moss, R. A.; Mallon, C. B.; Ho, C.-T. J. Am. Chem. Soc. 1977, 99, 4105–4110. doi:10.1021/ja00454a032 |
| 81. | Bonge, H. T.; Hansen, T. J. Org. Chem. 2010, 75, 2309–2320. doi:10.1021/jo100113b |
| 82. | Bonge, H. T.; Hansen, T. Eur. J. Org. Chem. 2010, 4355–4359. doi:10.1002/ejoc.201000531 |
| 83. | Bonge, H. T.; Hansen, T. Tetrahedron Lett. 2010, 51, 5378–5381. doi:10.1016/j.tetlet.2010.07.116 |
| 84. | Bonge, H. T.; Hansen, T. Pure Appl. Chem. 2011, 83, 565–575. doi:10.1351/PAC-CON-10-10-18 |
| 53. | Johansson, N. G. Acta Chem. Scand. 1973, 27, 1417–1420. doi:10.3891/acta.chem.scand.27-1417 |
| 54. | Åkermark, B.; Byström, S.; Florin, E.; Johansson, N.-G.; Lagerlund, I. Acta Chem. Scand., Ser. B 1974, 28, 375–376. doi:10.3891/acta.chem.scand.28b-0375 |
| 58. | Corey, E. J.; Felix, A. M. J. Am. Chem. Soc. 1965, 87, 2518–2519. doi:10.1021/ja01089a055 |
| 85. | Johansson, N. G.; Åkermark, B. Tetrahedron Lett. 1971, 12, 4785–4786. doi:10.1016/S0040-4039(01)97615-4 |
| 46. | Kagan, H. B.; Basselier, J.-J.; Luche, J.-L. Tetrahedron Lett. 1964, 5, 941–948. doi:10.1016/S0040-4039(00)90411-8 |
| 47. | Barrow, K. D.; Spotswood, T. M. Tetrahedron Lett. 1965, 6, 3325–3335. doi:10.1016/S0040-4039(01)89203-0 |
| 48. | McMillan, I.; Stoodley, R. J. Tetrahedron Lett. 1966, 7, 1205–1210. doi:10.1016/S0040-4039(00)72394-X |
| 49. | Decazes, J.; Luche, J. L.; Kagan, H. B. Tetrahedron Lett. 1970, 11, 3661–3664. doi:10.1016/S0040-4039(01)98555-7 |
| 44. | Davies, H. M. L.; Yang, J. Adv. Synth. Catal. 2003, 345, 1133–1138. doi:10.1002/adsc.200303089 |
| 77. | Davies, H. M. L.; Venkataramani, C.; Hansen, T.; Hopper, D. W. J. Am. Chem. Soc. 2003, 125, 6462–6468. doi:10.1021/ja0290072 |
| 50. | Rando, R. R. J. Am. Chem. Soc. 1970, 92, 6706–6707. doi:10.1021/ja00725a091 |
| 51. | Rando, R. R. J. Am. Chem. Soc. 1972, 94, 1629–1631. doi:10.1021/ja00760a033 |
| 52. | Tomioka, H.; Kondo, M.; Izawa, Y. J. Org. Chem. 1981, 46, 1090–1094. doi:10.1021/jo00319a010 |
| 76. | Pirrung, M. C.; Morehead, A. T., Jr. J. Am. Chem. Soc. 1994, 116, 8991–9000. doi:10.1021/ja00099a017 |
| 78. | Doyle, M. P.; Ren, T. The Influence of Ligands on Dirhodium(II) on Reactivity and Selectivity in Metal Carbene Reactions. In Progress in Inorganic Chemistry; Karlin, K. D., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001; pp 113–168. |
| 79. | Lloret, J.; Carbo, J. J.; Bo, C.; Lledos, A.; Perez-Prieto, J. Organometallics 2008, 27, 2873–2876. doi:10.1021/om800089k |
| 80. | Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o |
| 81. | Bonge, H. T.; Hansen, T. J. Org. Chem. 2010, 75, 2309–2320. doi:10.1021/jo100113b |
© 2013 Kaupang and Bonge-Hansen; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)