Abstract
The synthesis of 2-tetralones through the cyclization of δ-aryl-β-dicarbonyl substrates by using CAN is described. Appropriately functionalized aromatic substrates undergo intramolecular cyclizations generating 2-tetralone derivatives in moderate to good yields. DFT computational studies indicate that successful formation of 2-tetralones from δ-aryl-β-dicarbonyl radicals is dependent on the stability of the subsequent cyclohexadienyl radical intermediates. Furthermore, DFT computational studies were used to rationalize the observed site selectivity in the 2-tetralone products.

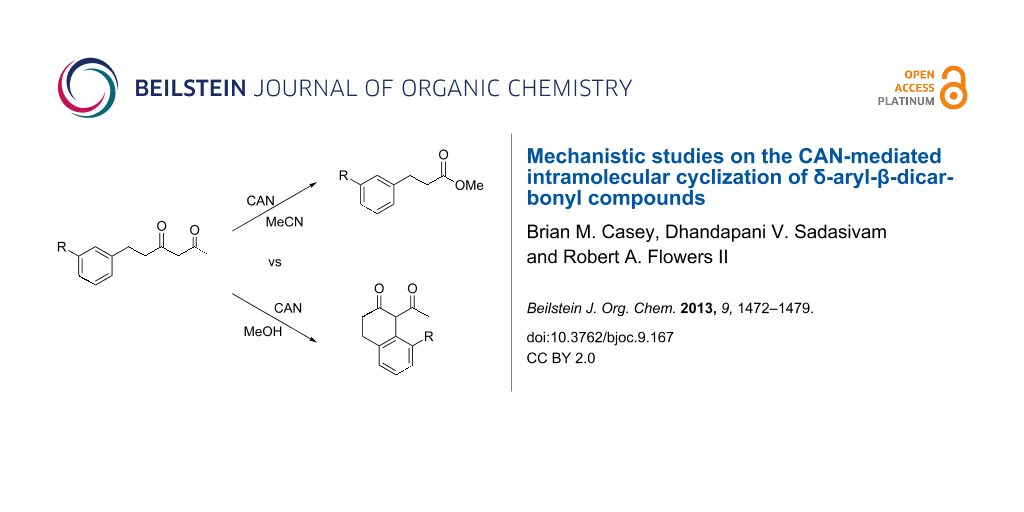
Graphical Abstract
Introduction
2-Tetralones are important intermediates or components of several natural products and biologically relevant molecules [1-7]. They are typically synthesized through transition-metal-mediated processes or preformed tetralin or naphthyl precursors [8,9]. Radical approaches can also be used to synthesize substituted 2-tetralones, and single-electron oxidations employing δ-aryl-β-dicarbonyl compounds have been carried out on arenes containing pendant β-ketoesters [10,11]. When Mn(III)-based oxidants are employed, secondary oxidations of the 2-tetralone products can occur [10,11].
Cerium(IV) ammonium nitrate (CAN) is a versatile, inexpensive and nontoxic single-electron oxidizing reagent commonly used in organic synthesis [12-14]. In a previous study, we showed that when 6-phenyl-2,4-hexanedione (1a) is oxidized by CAN in MeCN in the absence of a radicophile, 3-phenylpropionic acid (2a*) is obtained exclusively over the cyclized 2-tetralone product 2a (Scheme 1) [15]. Using this method, a variety of 1,3-dicarbonyl compounds can be mildly converted to carboxylic acids in moderate to excellent yields. In follow up studies, we found that 2-tetralone 2a could be obtained as the major product when 1a was oxidized by CAN in MeOH [16]. In addition, when 2.2 equivalents of CAN were employed, no products of secondary oxidations were obtained. Based on this observation, the single-electron oxidations of a variety of δ-aryl-β-dicarbonyl substrates with CAN in MeOH were performed to determine the scope of the reaction. DFT calculations were used to rationalize the observed impact of substitution on the δ-aryl ring on cyclization. The results of the synthetic and computational studies are presented herein.
![[1860-5397-9-167-i1]](/bjoc/content/inline/1860-5397-9-167-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Oxidative conversion of 1,3-dicarbonyl compounds to carboxylic acids with CAN.
Scheme 1: Oxidative conversion of 1,3-dicarbonyl compounds to carboxylic acids with CAN.
Results and Discussion
In an initial experiment, compound 1a was treated with 2.2 equivalents of CAN in MeOH producing 2-tetralone 2a in a 73% yield. To examine the breadth of this method, a series of δ-aryl-β-dicarbonyl substrates was prepared by a previously reported procedure [17]. As shown in Table 1, the intramolecular cyclization of δ-aryl-β-diketones with unsubstituted aryl rings (Table 1, entries 1 and 3) afforded 2-tetralone products in moderate to good yields. Additionally, cyclization of the β-ketoester 1b proceeded efficiently, generating 2b in an 85% yield.
Table 1: CAN-mediated oxidation of δ-aryl-β-dicarbonyl compounds in MeOHa.
| Entry | Substrate | Productb | Yield (%)c |
|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-167-i3.svg?max-width=637&scale=1.0)
1a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-167-i4.svg?max-width=637&scale=1.0)
2a |
73 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-167-i5.svg?max-width=637&scale=1.0)
1b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-167-i6.svg?max-width=637&scale=1.0)
2b |
85 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-167-i7.svg?max-width=637&scale=1.0)
1c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-167-i8.svg?max-width=637&scale=1.0)
2c |
59 |
aReaction conditions: 1 equiv δ-aryl-β-dicarbonyl, 2.2 equiv CAN, MeOH, rt, N2, 4 h. bProducts exist predominantly in the enol form by 1H NMR. cIsolated yield.
Previous work by Rickards and co-workers on a related system reported strong electronic effects when electron-donating substituents were incorporated onto the δ-aryl ring of the starting material [10,11]. To probe the impact of electron density of the δ-aryl ring on intramolecular cyclization, several substrates with either electron-donating or electron-withdrawing groups were synthesized and subjected to our reaction conditions. The results of these experiments are summarized in Table 2. As shown in Table 2, entry 1, dimethoxylated substrate 1d oxidatively cyclized to the 2-tetralone derivative in a 76% yield. However, when only one methoxy group was incorporated onto the δ-aryl ring, the expected 2-tetralone derivative was obtained only when the methoxy group was at the meta position (Table 2, entry 4). For substrates 1e and 1f with the methoxy group at either the ortho or para position, respectively, methyl esters 2e and 2f (Table 2, entries 2 and 3) were the major products. Additionally, intramolecular cyclizations with electron-deficient δ-aryl rings (Table 2, entries 5 and 6) did not occur, and oxidation of 1h and 1i instead favored the formation of methyl esters 2h and 2i. Finally, the tricyclic product 2j was generated in an isolated yield of 61% when substrate 1j was oxidized. It is important to note that products 2g and 2j were isolated as single isomers. Moreover, the isomers formed from these reactions result from cyclization occurring at the more hindered carbon atom of the δ-aryl ring. This observed site selectivity is consistent with previous research by both MacMillan and Nicolaou on the α-arylation of aldehydes through organo-SOMO activation [18-21]. The preferential formation of these isomers will be discussed below.
Table 2: Impact of ring substituents on the CAN-mediated oxidation of δ-aryl-β-dicarbonyl compounds in MeOHa.
| Entry | Substrate | Productb | Yield (%)c |
|---|---|---|---|
| 1 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-167-i9.svg?max-width=637&scale=1.0)
1d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-167-i10.svg?max-width=637&scale=1.0)
2d |
76 |
| 2 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-167-i11.svg?max-width=637&scale=1.0)
1e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-167-i12.svg?max-width=637&scale=1.0)
2e |
–d |
| 3 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-167-i13.svg?max-width=637&scale=1.0)
1f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-167-i14.svg?max-width=637&scale=1.0)
2f |
–d |
| 4 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-167-i15.svg?max-width=637&scale=1.0)
1g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-167-i16.svg?max-width=637&scale=1.0)
2g |
83 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-167-i17.svg?max-width=637&scale=1.0)
1h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-167-i18.svg?max-width=637&scale=1.0)
2h |
–d |
| 6 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-167-i19.svg?max-width=637&scale=1.0)
1i |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-167-i20.svg?max-width=637&scale=1.0)
2i |
–d |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-167-i21.svg?max-width=637&scale=1.0)
1j |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-167-i22.svg?max-width=637&scale=1.0)
2j |
61 |
aReaction conditions: 1 equiv δ-aryl-β-dicarbonyl, 2.2 equiv CAN, MeOH, rt, N2, 4 h. bProducts exist predominantly in the enol form by 1H NMR. cIsolated yield. dGC data indicated that the major products (50–80% conversion) were the methyl esters. Attempts were not made to isolate the methyl esters.
While the nucleophilicity of alkyl radicals is well-documented [22-24], the radicals generated from β-dicarbonyl compounds have been shown to display more electrophilic character [25-27]. As a consequence, these radicals should favor coupling with more nucleophilic, electron-rich carbon centers. The observation that intramolecular cyclization did not occur in either of the electron-deficient substrates (compounds 1h and 1i) is consistent with electrophilic radical intermediates.
To obtain a better understanding of the impact of arene substitution on the intramolecular cyclization, DFT calculations were performed at UB3LYP/6-31G(d) level by using Gaussian 03/09 [28]. Previous research from our group has shown that the oxidation of the enol tautomer of a diketone initially forms a radical cation, which is deprotonated readily by MeOH to form the radical under the reaction conditions [15,16].
To probe the origin of the effect of substitution on cyclization, the key step of the reaction, namely the cyclization of the β-dicarbonyl radical onto the aromatic ring, was investigated. Energy barriers for the transition structures of the ortho cyclization leading to their corresponding product cyclohexadienyl radicals were determined.
All structures were fully optimized and identified as minima on potential energy surfaces with frequency calculations. Transition structures were identified with one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were performed to connect the transition structures to their respective minima on either side of the first-order saddle point. In some cases, the lowest energy structures obtained from IRCs were further optimized to obtain the minima.
For the aryl diketone radical, two conformers for the ketones were considered, one with the carbonyl groups syn to each other and the other with the carbonyl groups in the anti orientation. For every substrate, two transition structures were identified on the energy surface along the reaction coordinate. The first one is a rotational transition structure for the rotation around the C–C bond beta to the dicarbonyl to go from the minimum to a geometry from which an addition to the aromatic ring is viable. The second transition structure corresponds to the addition of the radical intermediate to the aromatic ring.
Recently, Houk, MacMillan and co-workers showed that for the organo-SOMO-catalyzed oxidative α-arylation of aldehydes, the preference for the attack of the intermediate enamine radical cation on the substituted aromatic ring leading to ortho/para cyclization depends on the greater stabilization of the intermediate cyclohexadienyl radical [19]. The oxidative cyclization of δ-aryl-β-dicarbonyl substrates should proceed through a similar intermediate. As a result, the same rationale can be applied here. While MacMillan and Houk performed detailed computational studies to explain the site-selective radical cyclizations with m-methoxylated rings, the selectivity of naphthyl substrates was not investigated.
The energy values for the cyclization of all substituents are given in Table 3, and their corresponding energy diagrams are given in Figure 1. For unsubstituted 1a’, the initial rotational transition structure TS1a’ has a barrier of 2.5 kcal/mol, and the corresponding radical intermediate 2a’ is 0.6 kcal/mol above 1a’. For the radical addition to the aromatic ring, the energy barrier is 15.8 kcal/mol, and the product cyclohexadienyl radical 3a’ is 8.8 kcal/mol higher in energy. The syn isomer of 1a’ is 4.5 kcal/mol higher in energy compared to the anti isomer (see Table S2 in Supporting Information File 1). Only the energies of the anti isomers are discussed here.
Table 3: Energies (R. E. kcal/mol) of calculated structures. Energies are relative to the open form of the radical.
| R. E.a | R. E. + ZPVEb | low frequencyc | |
|---|---|---|---|
| 1a' | 0.0 | 0.0 | 21.4 |
| TS1a' | 2.3 | 2.5 | 46.7i |
| 2a' | 0.2 | 0.6 | 19.7 |
| TS2a' | 15.0 | 15.8 | 477.2i |
| 3a' | 7.7 | 8.8 | 35.4 |
| 1g' | 0.0 | 0.0 | 13.30 |
| TS1g' | 2.4 | 2.5 | 41.8i |
| 2g' | 0.4 | 0.7 | 16.6 |
| TS2g' | 11.0 | 12.0 | 446.3i |
| 3g' | 4.2 | 5.7 | 37.5 |
| 1g'' | 0.0 | 0.0 | 13.3 |
| TS1g" | 2.3 | 2.4 | 45.0i |
| 2g" | 0.3 | 0.6 | 13.5 |
| TS2g" | 13.1 | 13.9 | 437.5i |
| 3g" | 7.3 | 8.5 | 34.2 |
| 1f' | 0.0 | 0.0 | 21.3 |
| TS1f' | 2.4 | 2.4 | 42.3i |
| 2f' | 0.2 | 0.5 | 15.9 |
| TS2f' | 15.8 | 16.4 | 473.0i |
| 3f' | 9.1 | 10.0 | 36.1 |
| 1h' | 0.0 | 0.0 | 15.1 |
| TS1h' | 2.3 | 2.4 | 40.3i |
| 2h' | 0.2 | 0.4 | 19.1 |
| TS2h' | 16.2 | 16.9 | 489.8i |
| 3h' | 8.8 | 10.2 | 36.2 |
| 1j' | 0.0 | 0.0 | 17.45 |
| TS1j' | 2.4 | 2.5 | 42.1i |
| 2j' | 0.2 | 0.5 | 16.5 |
| TS2j' | 11.8 | 12.6 | 445.9i |
| 3j' | 2.4 | 4.0 | 34.9 |
| 1j" | 0.0 | 0.0 | 17.5 |
| TS1j" | 2.2 | 2.3 | 41.3i |
| 2j" | 0.3 | 0.6 | 11.5 |
| TS2j" | 13.4 | 14.1 | 463.8i |
| 3j" | 4.9 | 6.2 | 35.7 |
aUB3LYP/6-31G(d) geometry optimized. bFrom (a) with unscaled zero-point vibrational energy (ZPVE) corrections. cLow or imaginary frequencies (cm−1).
![[1860-5397-9-167-1]](/bjoc/content/figures/1860-5397-9-167-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Energy diagram for the unsubstituted arene with the carbonyl groups anti to each other. For TS1a’ the dihedral angle between atoms abcd = 6.4°. *The energy values for 1a’, 1g’, 1h’ and 1j’ are shown. 1g”, 1f’ and 1j” were not included for clarity.
Figure 1: Energy diagram for the unsubstituted arene with the carbonyl groups anti to each other. For TS1a’ t...
In the case of the electron-rich m-methoxy substituent 1g’, the energy barrier for the cyclization is 12.0 kcal/mol, which is approximately 4 kcal/mol lower than TS2a’. Due to the electrophilic character of the diketo radical intermediate, electron-rich systems are expected to favor cyclization. Similarly, the cyclized cyclohexadienyl radical 3g’ is approximately 3.0 kcal/mol more stable compared to 3a’. The stability of the cyclized radical is reflected in the activation barrier, and rotational transition structure TS1g’ has a barrier of 2.5 kcal/mol. For the electron-withdrawing chloro-substituted arene 1h’, the energy barrier for the cyclization on the aromatic ring is 16.9 kcal/mol, nearly 5 kcal/mol higher compared to the m-methoxy substituent. The corresponding cyclohexadienyl radical 3h’ is 10.2 kcal/mol higher in energy compared to the parent β-dicarbonyl radical 1h’. The rotational transition structure has an energy barrier of 2.4 kcal/mol, which is similar to other radical intermediates. The higher barrier for cyclization is consistent with synthetic data showing that no cyclized product is obtained in this case. In the case of the naphthyl system (1j’), the energy barrier for the cyclization is 12.6 kcal/mol. The cyclized product radical 3j’ is only 4.0 kcal/mol higher in energy when compared to the parent aryl diketone radical, and the rotational transition structure has a comparable barrier of 2.5 kcal/mol.
While it is evident why electron-rich systems should more readily cyclize, the observed site selectivity in products 2g and 2j warranted further investigation. For these substrates, there are two possible sites of ortho cyclization (Figure 2). For the m-methoxy substrate 1g, the energy barrier for the cyclization (TS2g’’), which would lead to product 2g”, is 1.9 kcal/mol higher relative to the other ortho cyclization (TS2g’). This finding is consistent with the synthetic observation that 3g’ is the only product observed. A similar trend was observed for the 2-naphthyl substrate 1j. The barrier for TS2j”, which would form anthracene-derived product 2j”, is 1.5 kcal/mol higher than TS2j’ and provides a rationale for the selective formation of the phenanthrene-derived product (2j).
![[1860-5397-9-167-2]](/bjoc/content/figures/1860-5397-9-167-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Possible products from the ortho cyclization of 1g and 1j.
Figure 2: Possible products from the ortho cyclization of 1g and 1j.
Finally, in order to better understand how the position of the methoxy group on the δ-aryl ring affects the formation of the β-tetralone product, the energies of the intermediates for the p-methoxy substrate 1f were calculated. The energy barrier for the cyclization (TS2f’) is 16.4 kcal/mol, 4.4 kcal/mol higher than the corresponding TS2g’. Furthermore, the product cyclohexadienyl radical 3f’ is 10.0 kcal/mol less stable than the parent 1f’, a value very close to the 10.2 kcal/mol for the electron-poor m-chloro substrate 3h’. Taken together, these data support the experimental observations that oxidative cyclization occurs only for the methoxy substrate substituted at the meta position.
Overall the DFT calculations show that the origin of the reactivity as well as the selectivity in these reactions depends on the stability of the product cyclohexadienyl radical, which is reflected in the activation barriers (TS2’s) for the cyclization, consistent with previous studies of MacMillan and Houk [19]. The m-methoxy substituent on the aromatic ring provides the lowest barrier for cyclization, and the corresponding cyclized cyclohexadienyl radical is more stable. Conversely, the electron-withdrawing m-chloro substituent has the highest barrier among the systems studied and leads to the least stable cyclohexadienyl radical. For the unsubstituted arene substrate, the energy barrier to cyclization as well as the energy of the cyclohexadienyl radical intermediate falls between the calculated values for the m-chloro and m-methoxy systems. For the 2-naphthyl system, delocalization of the cyclized radical intermediate results in a more stable product and hence a lower barrier providing a pathway to formation of phenanthrene-derived 2j.
In a previous study by our research group, the rates of oxidation of several β-diketones and their related silyl enol ethers by CAN and the more lipophilic ceric tetra-n-butylammonium nitrate (CTAN) were measured in MeOH, MeCN and CH2Cl2 by using stopped-flow spectrophotometry [16]. In these experiments, initial oxidation of substrates generated radical cation intermediates. The rates of formation and subsequent decay of these radical cations were measured in all three solvents. The results from these studies [15,16] provide two important insights into the mechanism of the oxidation of δ-aryl-β-dicarbonyl compounds in MeOH. First, MeOH is intimately involved in the decay of the initial radical cation through solvent-assisted deprotonation. Second, intramolecular cyclization of 1a’ occurs after the rate-limiting step of the reaction.
Based on the synthetic and computational data presented herein and findings from previous studies, the mechanism in Scheme 2 is proposed for the oxidation of δ-aryl-β-dicarbonyl compounds in MeOH with CAN. Initial oxidation of the enol tautomer (4’) by CAN produces protonated radical 5. Intermediate 5 is deprotonated by MeOH to radical species 6. When the radical contains a δ-aryl group with an appropriate substitution at the meta position, path A is followed. Intramolecular cyclization of 7 occurs through radical addition to the aromatic ring forming intermediate 8. As demonstrated by the oxidation of substrate 1j, intramolecular cyclization of this radical occurs at the more electron-rich carbon atom of the δ-aryl rings. A second equivalent of CAN oxidizes 8 to cation 9. Rearomatization through deprotonation of intermediate 9 yields the 2-tetralone derivative 10. Conversely, when the δ-aryl ring has electron-withdrawing substituents (Table 2, entries 5 and 6), the reaction follows path B [16,29-31].
![[1860-5397-9-167-i2]](/bjoc/content/inline/1860-5397-9-167-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the conversion of δ-aryl-β-dicarbonyl compounds to β-tetralones (path A) and methyl esters (path B).
Scheme 2: Proposed mechanism for the conversion of δ-aryl-β-dicarbonyl compounds to β-tetralones (path A) and...
Conclusion
A protocol for the conversion of δ-aryl-β-tetralones using CAN has been developed. The Ce(IV)-mediated synthesis of 2-tetralones has short reaction times and affords the desired products in moderate to very good yields under mild conditions. While 2-tetralones were not generated for all substrates, cyclization does occur for the unsubstituted arene 1a suggesting that the electrophilicity of the radical provides some driving force for the cyclization. The DFT computational studies indicated that the formation of 2-tetralones from the cyclization of δ-aryl-β-dicarbonyl radicals is dependent on the stability of the product cyclohexadienyl radicals.
Experimental
General methods and materials. Methanol (MeOH) was degassed with argon and dried with activated 3 Å molecular sieves prior to use. THF was purified with a Pure Solv solvent purification system from Innovative Technology Inc. CAN was purchased commercially and used without further purification. The glassware was flame dried prior to use. Unless otherwise stated, reactions were performed under an inert atmosphere of nitrogen. Products were separated by using prepacked silica gel columns with a gradient elution of ether/hexanes in an automated CombiFlash® Rf system from Teledyne Isco, Inc. All new compounds were characterized by 1H NMR, 13C NMR, GC–MS, IR, and LC–HRMS. Known compounds were characterized by 1H NMR, 13C NMR and GC–MS. 1H NMR and 13C NMR spectra were recorded on a Bruker 500 MHz spectrometer. Mass spectra were obtained by using a HP 5890 series GC–MS instrument. A Satellite FTIR from Thermo-Mattson was used to obtain IR spectra. LC–HRMS data were recorded at the Mass Spectrometry Facility at Notre Dame University.
General procedure for the synthesis of δ-aryl-β-dicarbonyl compounds 1a–j. Sodium hydride (11 mmol) was suspended in 25 mL of THF and cooled to 0 °C. Next, 10 mmol of 2,4-pentanedione (or methyl acetoacetate for 1b) was added dropwise to the flask, evolving H2 gas and forming an opaque, white solution. After stirring for 10 min, 10.5 mmol of butyllithium was added dropwise forming a clear yellow solution, which was stirred for an additional 10 min. The appropriate organohalide (11 mmol) was dissolved in 2 mL of THF and rapidly injected into the reaction at 0 °C. The reaction mixture was warmed gradually to room temperature over 30 min. The reaction was slowly quenched with an HCl solution (2 mL of concentrated HCl in 5 mL H2O). The organic layer was separated, and the aqueous layer was washed three times with ether. The organic layers were combined, washed with brine, dried with MgSO4, filtered, and concentrated. The crude product was purified by automated flash chromatography.
General procedure for the oxidation of δ-aryl-β-dicarbonyl compounds with CAN in MeOH. CAN (1.1 mmol) was dissolved in 4 mL MeOH. This CAN solution was then added dropwise in 1 min to the δ-aryl-β-dicarbonyl compound (0.5 mmol), which was dissolved in 15 mL of MeOH. The reaction was stirred for 30 min. The solvent was then removed by rotary evaporation. Ice-cold H2O (15 mL) was poured into the reaction and extracted three times with ether. The organic layers were combined, dried with MgSO4, filtered, and concentrated. The crude product was purified by automated flash chromatography.
Supporting Information
| Supporting Information File 1: Characterization data for all compounds, copies of 1H and 13C NMR spectra of final products, computational details, absolute energies, and Cartesian coordinates of all optimized structures. | ||
| Format: PDF | Size: 5.5 MB | Download |
References
-
Taber, D. F.; Neubert, T. D.; Rheingold, A. L. J. Am. Chem. Soc. 2002, 124, 12416–12417. doi:10.1021/ja027882h
Return to citation in text: [1] -
Gemma, S.; Butini, S.; Fattorusso, C.; Fiorini, I.; Nacci, V.; Bellebaum, K.; McKissic, D.; Saxena, A.; Campiani, G. Tetrahedron 2003, 59, 87–93. doi:10.1016/S0040-4020(02)01449-7
Return to citation in text: [1] -
Goel, A.; Verma, D.; Pratap, R.; Taneja, G.; Hemberger, Y.; Knauer, M.; Raghunandan, R.; Maulik, P. R.; Ram, V. J.; Bringmann, G. Eur. J. Org. Chem. 2011, 2940–2947. doi:10.1002/ejoc.201001565
Return to citation in text: [1] -
Silveira, C. C.; Machado, A.; Braga, A. L.; Lenardão, E. J. Tetrahedron Lett. 2004, 45, 4077–4080. doi:10.1016/j.tetlet.2004.03.154
Return to citation in text: [1] -
Lucas, S.; Heim, R.; Ries, C.; Schewe, K. E.; Birk, B.; Hartmann, R. W. J. Med. Chem. 2008, 51, 8077–8087. doi:10.1021/jm800888q
Return to citation in text: [1] -
Nguyen, T. X.; Kobayashi, Y. J. Org. Chem. 2008, 73, 5536–5541. doi:10.1021/jo800793s
Return to citation in text: [1] -
Yao, B.; Ji, H.; Cao, Y.; Zhou, Y.; Zhu, J.; Lü, J.; Li, Y.; Chen, J.; Zheng, C.; Jiang, Y.; Liang, R.; Tang, H. J. Med. Chem. 2007, 50, 5293–5300. doi:10.1021/jm0701167
Return to citation in text: [1] -
Silveira, C. C.; Braga, A. L.; Kaufmanb, T. S.; Lenardão, E. J. Tetrahedron 2004, 60, 8295–8328. doi:10.1016/j.tet.2004.06.080
Return to citation in text: [1] -
Hon, Y.-S.; Devulapally, R. Tetrahedron Lett. 2009, 50, 2831–2834. doi:10.1016/j.tetlet.2009.03.185
Return to citation in text: [1] -
Citterio, A.; Pesce, L.; Sebastiano, R.; Santi, R. Synthesis 1990, 142–144. doi:10.1055/s-1990-26814
Return to citation in text: [1] [2] [3] -
Jamie, J. F.; Rickards, R. W. J. Chem. Soc., Perkin Trans. 1 1996, 2603–2613. doi:10.1039/P19960002603
Return to citation in text: [1] [2] [3] -
Nair, V.; Deepthi, A. Tetrahedron 2009, 65, 10745–10755. doi:10.1016/j.tet.2009.10.083
Return to citation in text: [1] -
Nair, V.; Deepthi, A. Chem. Rev. 2007, 107, 1862–1891. doi:10.1021/cr068408n
Return to citation in text: [1] -
Casey, B. M.; Eakin, C. A.; Flowers, R. A., II. Tetrahedron Lett. 2009, 50, 1264–1266. doi:10.1016/j.tetlet.2008.12.114
Return to citation in text: [1] -
Zhang, Y.; Jiao, J.; Flowers, R. A., II. J. Org. Chem. 2006, 71, 4516–4520. doi:10.1021/jo0602975
Return to citation in text: [1] [2] [3] -
Jiao, J.; Zhang, Y.; Devery, J. J., III; Xu, L.; Deng, J.; Flowers, R. A., II. J. Org. Chem. 2007, 72, 5486–5492. doi:10.1021/jo0625406
Return to citation in text: [1] [2] [3] [4] [5] -
Huckin, S. N.; Weiler, L. J. Am. Chem. Soc. 1974, 96, 1082–1087. doi:10.1021/ja00811a023
Return to citation in text: [1] -
Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902
Return to citation in text: [1] -
Um, J. M.; Gutierrez, O.; Schoenebeck, F.; Houk, K. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 6001–6005. doi:10.1021/ja9063074
Return to citation in text: [1] [2] [3] -
Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 2086–2087. doi:10.1021/ja809405c
Return to citation in text: [1] -
Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 6640. doi:10.1021/ja902682t
Return to citation in text: [1] -
Minisci, F. Recent aspects of homolytic aromatic substitutions. In Synthetic and Mechanistic Organic Chemistry; Minisci, F.; Hendrickson, J. B.; Wentrup, C., Eds.; Topics in Current Chemistry, Vol. 62; Springer: Berlin, Germany, 1976; pp 1–48. doi:10.1007/BFb0046046
Return to citation in text: [1] -
Citterio, A.; Arnoldi, A.; Minisci, F. J. Org. Chem. 1979, 44, 2674–2682. doi:10.1021/jo01329a017
Return to citation in text: [1] -
Citterio, A.; Minisci, F.; Porta, O.; Sesana, G. J. Am. Chem. Soc. 1977, 99, 7960–7968. doi:10.1021/ja00466a031
Return to citation in text: [1] -
Heiba, E.-A. I.; Dassau, R. M. J. Org. Chem. 1974, 39, 3456–3457. doi:10.1021/jo00937a052
Return to citation in text: [1] -
Fristad, W. E.; Hershberger, S. S. J. Org. Chem. 1985, 50, 1026–1031. doi:10.1021/jo00207a024
Return to citation in text: [1] -
Corey, E. J.; Kang, M. C. J. Am. Chem. Soc. 1984, 106, 5384–5385. doi:10.1021/ja00330a076
Return to citation in text: [1] -
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2010.
Return to citation in text: [1] -
Goswami, P.; Chowdhury, P. New J. Chem. 2000, 24, 955–957. doi:10.1039/b005908k
Return to citation in text: [1] -
Iranpoor, N.; Shekarriz, M. Bull. Chem. Soc. Jpn. 1999, 72, 455–458. doi:10.1246/bcsj.72.455
Return to citation in text: [1] -
Pan, W.-B.; Chang, F.-R.; Wei, L.-M.; Wu, M.-J.; Wu, Y.-C. Tetrahedron Lett. 2003, 44, 331–334. doi:10.1016/S0040-4039(02)02578-9
Return to citation in text: [1]
| 16. | Jiao, J.; Zhang, Y.; Devery, J. J., III; Xu, L.; Deng, J.; Flowers, R. A., II. J. Org. Chem. 2007, 72, 5486–5492. doi:10.1021/jo0625406 |
| 29. | Goswami, P.; Chowdhury, P. New J. Chem. 2000, 24, 955–957. doi:10.1039/b005908k |
| 30. | Iranpoor, N.; Shekarriz, M. Bull. Chem. Soc. Jpn. 1999, 72, 455–458. doi:10.1246/bcsj.72.455 |
| 31. | Pan, W.-B.; Chang, F.-R.; Wei, L.-M.; Wu, M.-J.; Wu, Y.-C. Tetrahedron Lett. 2003, 44, 331–334. doi:10.1016/S0040-4039(02)02578-9 |
| 16. | Jiao, J.; Zhang, Y.; Devery, J. J., III; Xu, L.; Deng, J.; Flowers, R. A., II. J. Org. Chem. 2007, 72, 5486–5492. doi:10.1021/jo0625406 |
| 15. | Zhang, Y.; Jiao, J.; Flowers, R. A., II. J. Org. Chem. 2006, 71, 4516–4520. doi:10.1021/jo0602975 |
| 16. | Jiao, J.; Zhang, Y.; Devery, J. J., III; Xu, L.; Deng, J.; Flowers, R. A., II. J. Org. Chem. 2007, 72, 5486–5492. doi:10.1021/jo0625406 |
| 1. | Taber, D. F.; Neubert, T. D.; Rheingold, A. L. J. Am. Chem. Soc. 2002, 124, 12416–12417. doi:10.1021/ja027882h |
| 2. | Gemma, S.; Butini, S.; Fattorusso, C.; Fiorini, I.; Nacci, V.; Bellebaum, K.; McKissic, D.; Saxena, A.; Campiani, G. Tetrahedron 2003, 59, 87–93. doi:10.1016/S0040-4020(02)01449-7 |
| 3. | Goel, A.; Verma, D.; Pratap, R.; Taneja, G.; Hemberger, Y.; Knauer, M.; Raghunandan, R.; Maulik, P. R.; Ram, V. J.; Bringmann, G. Eur. J. Org. Chem. 2011, 2940–2947. doi:10.1002/ejoc.201001565 |
| 4. | Silveira, C. C.; Machado, A.; Braga, A. L.; Lenardão, E. J. Tetrahedron Lett. 2004, 45, 4077–4080. doi:10.1016/j.tetlet.2004.03.154 |
| 5. | Lucas, S.; Heim, R.; Ries, C.; Schewe, K. E.; Birk, B.; Hartmann, R. W. J. Med. Chem. 2008, 51, 8077–8087. doi:10.1021/jm800888q |
| 6. | Nguyen, T. X.; Kobayashi, Y. J. Org. Chem. 2008, 73, 5536–5541. doi:10.1021/jo800793s |
| 7. | Yao, B.; Ji, H.; Cao, Y.; Zhou, Y.; Zhu, J.; Lü, J.; Li, Y.; Chen, J.; Zheng, C.; Jiang, Y.; Liang, R.; Tang, H. J. Med. Chem. 2007, 50, 5293–5300. doi:10.1021/jm0701167 |
| 12. | Nair, V.; Deepthi, A. Tetrahedron 2009, 65, 10745–10755. doi:10.1016/j.tet.2009.10.083 |
| 13. | Nair, V.; Deepthi, A. Chem. Rev. 2007, 107, 1862–1891. doi:10.1021/cr068408n |
| 14. | Casey, B. M.; Eakin, C. A.; Flowers, R. A., II. Tetrahedron Lett. 2009, 50, 1264–1266. doi:10.1016/j.tetlet.2008.12.114 |
| 19. | Um, J. M.; Gutierrez, O.; Schoenebeck, F.; Houk, K. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 6001–6005. doi:10.1021/ja9063074 |
| 10. | Citterio, A.; Pesce, L.; Sebastiano, R.; Santi, R. Synthesis 1990, 142–144. doi:10.1055/s-1990-26814 |
| 11. | Jamie, J. F.; Rickards, R. W. J. Chem. Soc., Perkin Trans. 1 1996, 2603–2613. doi:10.1039/P19960002603 |
| 19. | Um, J. M.; Gutierrez, O.; Schoenebeck, F.; Houk, K. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 6001–6005. doi:10.1021/ja9063074 |
| 10. | Citterio, A.; Pesce, L.; Sebastiano, R.; Santi, R. Synthesis 1990, 142–144. doi:10.1055/s-1990-26814 |
| 11. | Jamie, J. F.; Rickards, R. W. J. Chem. Soc., Perkin Trans. 1 1996, 2603–2613. doi:10.1039/P19960002603 |
| 8. | Silveira, C. C.; Braga, A. L.; Kaufmanb, T. S.; Lenardão, E. J. Tetrahedron 2004, 60, 8295–8328. doi:10.1016/j.tet.2004.06.080 |
| 9. | Hon, Y.-S.; Devulapally, R. Tetrahedron Lett. 2009, 50, 2831–2834. doi:10.1016/j.tetlet.2009.03.185 |
| 15. | Zhang, Y.; Jiao, J.; Flowers, R. A., II. J. Org. Chem. 2006, 71, 4516–4520. doi:10.1021/jo0602975 |
| 16. | Jiao, J.; Zhang, Y.; Devery, J. J., III; Xu, L.; Deng, J.; Flowers, R. A., II. J. Org. Chem. 2007, 72, 5486–5492. doi:10.1021/jo0625406 |
| 10. | Citterio, A.; Pesce, L.; Sebastiano, R.; Santi, R. Synthesis 1990, 142–144. doi:10.1055/s-1990-26814 |
| 11. | Jamie, J. F.; Rickards, R. W. J. Chem. Soc., Perkin Trans. 1 1996, 2603–2613. doi:10.1039/P19960002603 |
| 22. | Minisci, F. Recent aspects of homolytic aromatic substitutions. In Synthetic and Mechanistic Organic Chemistry; Minisci, F.; Hendrickson, J. B.; Wentrup, C., Eds.; Topics in Current Chemistry, Vol. 62; Springer: Berlin, Germany, 1976; pp 1–48. doi:10.1007/BFb0046046 |
| 23. | Citterio, A.; Arnoldi, A.; Minisci, F. J. Org. Chem. 1979, 44, 2674–2682. doi:10.1021/jo01329a017 |
| 24. | Citterio, A.; Minisci, F.; Porta, O.; Sesana, G. J. Am. Chem. Soc. 1977, 99, 7960–7968. doi:10.1021/ja00466a031 |
| 17. | Huckin, S. N.; Weiler, L. J. Am. Chem. Soc. 1974, 96, 1082–1087. doi:10.1021/ja00811a023 |
| 25. | Heiba, E.-A. I.; Dassau, R. M. J. Org. Chem. 1974, 39, 3456–3457. doi:10.1021/jo00937a052 |
| 26. | Fristad, W. E.; Hershberger, S. S. J. Org. Chem. 1985, 50, 1026–1031. doi:10.1021/jo00207a024 |
| 27. | Corey, E. J.; Kang, M. C. J. Am. Chem. Soc. 1984, 106, 5384–5385. doi:10.1021/ja00330a076 |
| 16. | Jiao, J.; Zhang, Y.; Devery, J. J., III; Xu, L.; Deng, J.; Flowers, R. A., II. J. Org. Chem. 2007, 72, 5486–5492. doi:10.1021/jo0625406 |
| 15. | Zhang, Y.; Jiao, J.; Flowers, R. A., II. J. Org. Chem. 2006, 71, 4516–4520. doi:10.1021/jo0602975 |
| 18. | Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902 |
| 19. | Um, J. M.; Gutierrez, O.; Schoenebeck, F.; Houk, K. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2010, 132, 6001–6005. doi:10.1021/ja9063074 |
| 20. | Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 2086–2087. doi:10.1021/ja809405c |
| 21. | Nicolaou, K. C.; Reingruber, R.; Sarlah, D.; Bräse, S. J. Am. Chem. Soc. 2009, 131, 6640. doi:10.1021/ja902682t |
© 2013 Casey et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)