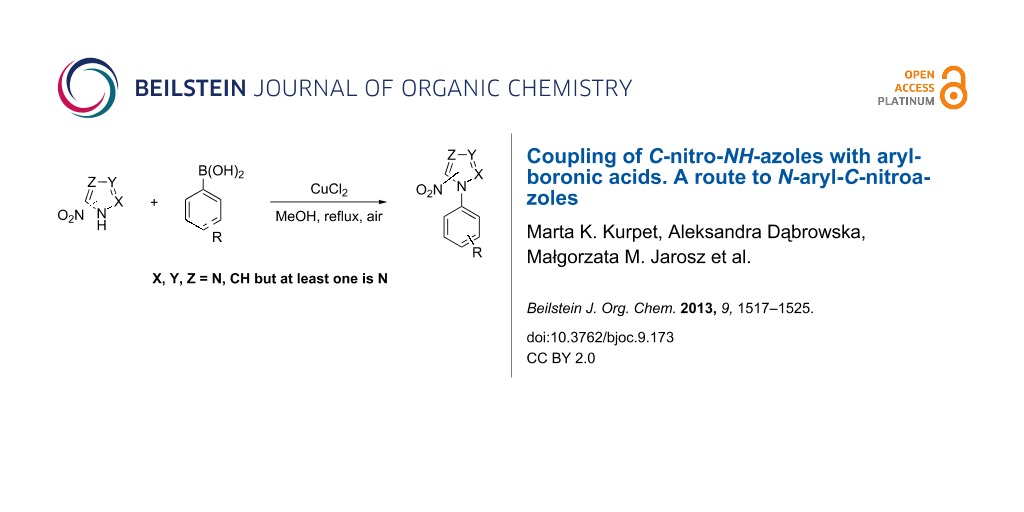
Abstract
A method for the synthesis of N-aryl-C-nitroazoles is presented. A coupling reaction between variously substituted arylboronic acids and 3(5)-nitro-1H-pyrazole catalyzed by copper salt has been carried out in methanol in the presence of sodium hydroxide to afford the desired N-aryl-C-nitroazoles in good yields. This synthetic route has also been successfully applied to obtain N-phenyl derivatives of 4-nitropyrazole, 2-nitroimidazole, 4(5)-nitroimidazole and 3-nitro-1,2,4-triazole.

Graphical Abstract
Introduction
The nitroazoles constitute a class of compounds with a broad spectrum of useful properties. They have found applications in agrochemicals as plant-growth regulators [1], herbicides or insecticides [2], in veterinary science [3], and as propellants and precursors of energetic materials [4]. Special attention has been paid to the development of their application in medicine where so far they are used as antiprotozoal, antifungal, and antibacterial drugs [5,6], as hypoxic cell sensitizers in radiation therapy of cancer [7], or as antiphlogistic drugs [8]. Many derivatives of C-nitroazoles, particularly N-alkylnitroimidazoles being the most often repeating subunit in biologically active compounds, suffer from mutagenic and carcinogenic properties [9]. Therefore the efforts of several research groups are focused on finding new compounds bearing C-nitroazole rings in their structure but exhibiting a less toxic effect.
Recent studies on biologically active derivatives of nitroazoles led to the synthesis and wide investigation of antibacterial and antiprotozoal 4-nitroimidazoles functionalized with an aryl substituent on the ring nitrogen atom. Many of these compounds exhibit a potent and selective anti-trypanosomal activity with relatively low mutagenic characteristics as well as genotoxic risk [10] and show promising antitubercular properties [11]. This opens an opportunity to look for new N-aryl-C-nitroazoles with the desired biological activity. Very recently, we reviewed the syntheses of N-phenyl-C-nitroazoles [12], and syntheses of their derivatives will be reviewed soon. The range of methods of N-aryl-C-nitroazoles synthesis includes nitration [13], ring closure [14,15], degenerated ANRORC reactions [16], transformation of other cyclic molecules [17], oxidation of aminoazoles [18], and introduction of an aryl substituent at the ring nitrogen atom in starting nitroazole [19,20]. Many of these methods suffer due to the inconvenient conditions of reactions, lack of selectivity, or low yields of the products. Therefore, there is a need for the development of a new, simple synthetic route, applicable to a wide range of C-nitro-NH-azoles.
Since 1998 when Chan and Lam [21,22] described a novel method for the formation of C–N bonds through the copper-catalyzed coupling of NH-containing substrates such as amines, amides, imides or heterocycles with boronic acids, the method has become a powerful synthetic route for N-arylation. Many investigations concerning the influence of the amount and kind of a catalyst, type of solvent, temperature, the presence of a base, and other reaction conditions, have carried out [23-27].
We were interested in developing an easy and effective method for the synthesis of N-aryl-C-nitroazoles that would allow us to obtain a broad range of the desired products bearing both chemically and electronically varying substituents on the benzene ring. The presence of the nitro group (a strong electron-withdrawing substituent) in the azole entails the adjustment of the conditions of N-arylation reaction.
Besides a very recently published approach involving N-arylation of nitroazoles with diaryliodonium salts [20], already described methods utilize arylboronic acids and substituted C-nitroazole coupling, applying the conditions described for the Chan–Lam reaction. This involves the use of chlorinated solvents, pyridine as base, and prolonged reaction times to obtain high yields of the product [28,29]. No detailed investigation on the influence of the conditions on the yield of the product has been presented.
Results and Discussion
The group of products that we mainly describe is the 1-aryl derivatives of 3(5)-nitro-1H-pyrazole (1a). This is still a barely investigated group of compounds that can have attractive properties. Some recent reports describe them as new glucagon receptor antagonists [28] and compounds having insectidal properties [30]. They are a substructure of compounds being inhibitors of cannabinoid receptor 1 [31], they can be used as intermediate products, e.g., in the synthesis of compounds for inhibiting phosphate transport [32], and are described as being useful for the treatment of cognitive-deficit-associated psychiatric, neurodegenerative and neurological disorders [33].
Only four papers describe the synthesis of 3-nitro-1-phenyl-1H-pyrazole. It may be a product of oxidation of 3-amino-1-phenyl-1H-pyrazole [18], a product of coupling of diaryliodonium salts with 3-nitro-1H-pyrazole [20], a product of Ullman phenylation of 3-nitro-1H-pyrazole [34], or a byproduct in the synthesis of 1-phenyl-4-amino-5-methylaminopyridazin-6-one [17] (Scheme 1).
![[1860-5397-9-173-i1]](/bjoc/content/inline/1860-5397-9-173-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Methods of synthesis of 3-nitro-1-phenyl-1H-pyrazole (3a) described in the literature.
Scheme 1: Methods of synthesis of 3-nitro-1-phenyl-1H-pyrazole (3a) described in the literature.
Although these works describe 3-nitro-1-phenyl-1H-pyrazole (3a) as the product of the reaction, different physical properties are specified below.
Despite, we obtained 3-nitro-1-phenyl-1H-pyrazole (3a), for which the spectra determined in chloroform-d were in excellent agreement with those presented by Chertkov et al. for their product [20], the melting points differed from each other by almost 30 °C.
The melting point of the product synthesized by us is 98–99 °C, which is consistent with results obtained by Coburn [18], who oxidized 3-amino-1-phenyl-1H-pyrazole (Scheme 1, method A), while Chertkov et al. determined a value of 127 °C (Scheme 1, method B), which on the other hand fits with the data published by Predvoditeleva [17] (Scheme 1, method D).
The presence of tautomeric forms of 3(5)-nitro-1H-pyrazole (1a) and the discrepancy in the melting point of our product and the recently described one, led us to look carefully into its structure and to investigate the possibility of formation of other isomers.
The analysis of 13C NMR spectra of our products shows that there are characteristic signals, which can be assigned to carbon atoms in the 3-nitro-pyrazole rings: about 103–105 ppm for C4-Py, 129–132 ppm for C5-Py and 156–157 ppm for C3-Py. These results are consistent with the analysis of regioisomers of 1-substituted-C-nitropyrazoles presented by Larina and Lopyrev in their review on nitroazoles [35]. Table 1 contains exemplified structures of 3-nitro-, 4-nitro- and 5-nitropyrazole derivatives and shows the differences in chemical shifts of carbon atoms in particular isomers. The examples do not include the 1-aryl substituent, which is present in our product, but general trends in chemical shifts can be observed. The C–NO2 signal in 5-nitropyrazole is shifted by about 10 ppm downfield in comparison to the C–NO2 signal in 3-nitropyrazole, what agrees with spectra recorded by us.
Table 1: 13C NMR chemical shifts (ppm) of C-nitropyrazoles [35].
| Substituents in nitropyrazole ring | 13C NMR chemical shifts (ppm) for nitropyrazoles | ||||||
|---|---|---|---|---|---|---|---|
| R1 | R3 | R4 | R5 | C3 | C4 | C5 | solvent |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-173-i5.svg?max-width=637&scale=1.0)
|
|||||||
| H | – | H | H | 155.70 | 103.24 | 132.80 | CD3OD |
| CH3 | – | H | H | 154.90 | 102.70 | 134.50 | DMSO-d6 |
| NH2 | – | H | H | 152.96 | 102.17 | 132.80 | DMSO-d6 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-173-i6.svg?max-width=637&scale=1.0)
|
|||||||
| H | H | – | H | 132.41 | 136.00 | 132.44 | DMSO-d6 |
| CH3 | H | – | H | 135.00 | 134.90 | 130.60 | DMSO-d6 |
| NH2 | H | – | H | 132.96 | 133.33 | 128.32 | DMSO-d6 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-173-i7.svg?max-width=637&scale=1.0)
|
|||||||
| CH3 | H | H | – | 137.60 | 106.30 | 145.80 | DMSO-d6 |
| NH2 | H | H | – | 133.60 | 104.72 | 142.26 | DMSO-d6 |
Additionally, in order to confirm the structure we used X-ray analysis for one of our products. The analyzed crystal of 3-nitro-1-[4-(trifluoromethoxy)phenyl]-3-nitro-1H-pyrazole (3k) was a monocrystal recrystallized from diethyl ether (Figure 1). It forms a monoclinic unit cell with two symmetry-related pairs of molecules. The molecule is relatively flat, with the benzene ring slightly twisted out from the pyrazole plane 19.60° (27), while the nitro-substituent is almost coplanar with pyrazole root 8.06° (90). Such a structure enables efficient overlapping of π orbitals resulting in high conjugation.
![[1860-5397-9-173-1]](/bjoc/content/figures/1860-5397-9-173-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray structure of 3-nitro-1-[4-(trifluoromethoxy)phenyl]-3-nitro-1H-pyrazole (3k) with 60% probability ellipsoids.
Figure 1: X-ray structure of 3-nitro-1-[4-(trifluoromethoxy)phenyl]-3-nitro-1H-pyrazole (3k) with 60% probabi...
Based on the agreement of our NMR spectra with those presented and thoroughly analyzed by Chertkov et al., as well as on our X-ray analysis, the substitution of the nitro-group in the 3-position of the 1H-pyrazole moiety was confirmed.
Our screening of the conditions involved the factors influencing the chemical yield of reaction, including the solvent/base system, the type of catalyst, and the stoichiometry of the reagents. 3-Nitro-1H-pyrazole (1a) and phenylboronic acid (2a) were used as model substrates to optimize the reaction conditions (Scheme 2).
![[1860-5397-9-173-i2]](/bjoc/content/inline/1860-5397-9-173-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cross coupling of 3-nitro-1H-pyrazole (1a) with phenylboronic acid (2a).
Scheme 2: Cross coupling of 3-nitro-1H-pyrazole (1a) with phenylboronic acid (2a).
At the very beginning we decided to perform several experiments that would allow us to establish the necessity of the presence of a catalyst, air and a base in the reaction system (Table 2). The initial conditions of the reaction were based on those published by Lan et al. [24] for coupling imidazole with phenylboronic acid. Most reports on the Chan–Lam coupling reaction underline the demand of air introduction into the reaction mixture to provide high yields of the products [22,24,36,37]. The plausible mechanism of this catalytic reaction was proposed by Evans [38] and described for N-nucleophiles by Collman [39]. It involves several steps: transmetallation of boronic acid with a catalyst, coordination of the azole molecule to the Cu(II) species followed by oxidation of copper(II) into copper(III) in the presence of oxygen, and then reductive elimination releasing the product and Cu(I) complex. A regeneration of the catalyst takes place in the presence of oxygen reproducing the Cu(II) cation.
Table 2: The influence of the presence of air, a base and a catalyst on the yield of 3-nitro-1-phenyl-1H-pyrazole (3a).a
| Entry | Catalyst | Base | Air | Yieldb |
|---|---|---|---|---|
| 1 | CuI | no | noe | 0% |
| 2c | CuI | no | + | trace |
| 3d | CuI | NaOH | noe | trace |
| 4c,d | CuI | NaOH | + | 64% |
| 5d | CuCl2 | NaOH | noe | 56% |
| 6c,d | CuCl2 | NaOH | + | 69% |
| 7c,d | – | NaOH | + | 0% |
aConditions: 2 mmol of 2a, 2.4 mmol of 1a, 10 mol % of catalyst, methanol, reflux. The progress of the reaction was controlled by TLC. bIsolated yields. cAir was bubbled through solution. dIn the presence of 2.4 mmol of NaOH. eNo bubbling of air through solution.
In the case of the application of Cu(I) salts, for which our preliminary studies were carried out, the presence of air proved to be obligatory. Attempts to perform the reaction without air resulted in no or only trace amounts of product being detected on the TLC plate (Table 2, entries 1 and 3). Our further experiments carried out in the presence of Cu(II) salt revealed that the air does not seem to be necessarily introduced for the reaction to take place (Table 2, entry 5). However, bubbling air through the reaction mixture improved the yield (Table 2, entry 6), indicating its possible impact on the catalytic cycle. These results are in agreement with those obtained by Strijdonck [40] who carried out copper-catalyzed coupling under anaerobic conditions. The mechanism of this reaction is still a matter of research. The reaction does not take place without a base (Table 2, entry 2) or in the absence of copper catalyst (Table 2, entry 7).
A parameter influencing chemical yields to a greater extent was the applied solvent/base system. Sodium hydroxide (pKa = 15.7) [41] and triethylamine (Et3N) (pKa = 10.9) [42] in dichloromethane, acetonitrile, dimethylformamide, tetrahydrofuran and methanol were investigated. Dichloromethane, being the most often reported solvent for coupling [21-23,36], turned out to be the least appropriate in the case of C-nitroazoles. Yields of the product obtained were then 3% and 8% in the presence of Et3N and NaOH, respectively (Table 3, entries 1,2). Even prolonged time did not improve the yield (Table 3, entry 3). Better results were obtained for more polar solvents (acetonitrile, dimethylformamide, tetrahydrofuran, methanol) independently on a base (Table 3, entries 4–11). The most efficient combinations were dimethylformamide/sodium hydroxide and methanol/sodium hydroxide. Methanol was the solvent of choice due to higher yields and convenience of workup and purification of the product.
Table 3: Influence of the solvent/base system on the yield of 3a.a
| Entry | Solvent | Base | Time [h] | Yieldb |
|---|---|---|---|---|
| 1 | CH2Cl2 | NaOH | 11 | 8 % |
| 2 | CH2Cl2 | Et3N | 10 | 3% |
| 3 | CH2Cl2 | NaOH | 20 | 8% |
| 4 | CH3CN | NaOH | 11 | 48% |
| 5 | CH3CN | Et3N | 11 | 40% |
| 6 | DMF | NaOH | 9 | 56% |
| 7 | DMF | Et3N | 11 | 40% |
| 8 | THF | NaOH | 10 | 48% |
| 9 | THF | Et3N | 11 | 37% |
| 10 | CH3OH | NaOH | 10 | 64% |
| 11 | CH3OH | Et3N | 11 | 26% |
| 12 | CH3OH | K2CO3 | 13 | 25% |
| 13 | CH3OH | DBU | 12 | 63% |
aConditions: 2 mmol of 2a, 2.4 mmol of 1a, 10 mol % of CuI, 2.4 mmol of base, methanol, reflux, air bubbled through solution. The progress of the reaction was controlled by TLC. bIsolated yields.
In order to complete the research on the solvent/base system, the coupling reaction was carried out in methanol in the presence of other bases such as potassium carbonate (pKa = 10.33) [43] and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKa = 12) [44] (Table 3, entries 12,13). Although the pKa values were determined in water, they are usually in good agreement in other polar solvents scales [45]. Not surprisingly, the differences in pKa values of the bases are reflected in the results obtained. Very strong bases (DBU, NaOH) are needed for the reaction to take place. Although the reaction with DBU yielded a similar amount of product as NaOH, the latter was chosen as being of lower toxicity, easier to process and environmentally friendly.
Next the catalytic activity of the copper salts was screened. Most known reports concerning N-arylation focus on Cu(OAc)2 salt [21,22], complexes of Cu(II) with different ligands [23,36], and heterocyclic copper-based catalysts [25,37]. Basically, easily available simple copper(I) and copper(II) compounds, mostly salts, were investigated. The yields obtained for Cu(I) salts (Table 4, entries 1–3) were slightly lower than those achieved for Cu(II) salts (Table 4, entries 5–8). However, the differences are small, and it can be assumed that under the reaction conditions applied the choice between Cu(I) and Cu(II) is not crucial, considering the yield of 3-nitro-1-phenyl-1H-pyrazole. Also the effect of the counterion seems to be insignificant. Among the investigated compounds only CuO was ineffective as a catalyst and gave only trace amounts of the product as detected on the TLC plate. This implies the need to consider another factor. The use of Cu(I) species obligates the introduction of air into the reaction mixture during the reaction. As our previous experiments confirmed, it is not required for Cu(II) salts, although it provides higher yields of the product.
Table 4: The influence of copper catalyst on the yield of 3a.a
| Entry | Catalyst | Yieldb |
|---|---|---|
| 1 | CuI | 63% |
| 2 | CuCl | 66% |
| 3 | CuBr | 58% |
| 4 | CuO | trace |
| 5 | CuCl2 | 69% |
| 6 | Cu(OAc)2·H2O | 66% |
| 7 | CuSO4 | 66% |
| 8 | Cu(acac)2 | 53% |
aConditions: 2 mmol of 2a, 2.4 mmol of 1a, 10% mol of a catalyst, 2.4 mmol of NaOH, methanol, reflux, 10 h, air bubbled through solution. The progress of the reaction was controlled by TLC. bIsolated yields.
It turned out that the ratio of reagents has an important influence on the chemical yield of 3-nitro-1-phenyl-1H-pyrazole. A ratio of arylboronic acid to 3-nitro-1H-pyrazole to the base of 2.6:1.6:1.6 seems to be optimal. This gave the highest (up to 82%) yield of the product. A series of experiments, when either an excess of azole or a base over other reagents was used, resulted in decreased yields. Nevertheless, the base should be used in stoichiometric proportion to the azole. Both increasing and decreasing the proportion led to lower yield of the product.
With an efficient N-arylating system for phenylboronic acid, we expanded the scope of this reaction by exploring a variety of boronic acids with different substitution patterns. The investigated compounds brought such substituents as F, Cl, CH3, OCH3, OCF3, and NO2 (Scheme 3). The results show that an electron-donating character of a substituent (Table 5, entries 7–10) allows higher yields of the products to be obtained than with electron-withdrawing groups (Table 5, entries 2–6 and entries 11–14).
![[1860-5397-9-173-i3]](/bjoc/content/inline/1860-5397-9-173-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cross coupling of 1a with arylboronic acids 2a–n.
Scheme 3: Cross coupling of 1a with arylboronic acids 2a–n.
Table 5: Coupling of 1a with arylboronic acids 2a–n.a
| Entry | Boronic acid | Product | Time [h] | Yieldb |
|---|---|---|---|---|
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-173-i8.svg?max-width=637&scale=1.0)
2a |
3a | 18 | 82% |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-173-i9.svg?max-width=637&scale=1.0)
2b |
3b | 11 | 64% |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-173-i10.svg?max-width=637&scale=1.0)
2c |
3c | 11 | 58% |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-173-i11.svg?max-width=637&scale=1.0)
2d |
3d | 25 | 50% |
| 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-173-i12.svg?max-width=637&scale=1.0)
2e |
3e | 16 | 67% |
| 6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-173-i13.svg?max-width=637&scale=1.0)
2f |
3f | 8 | 44% |
| 7 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-173-i14.svg?max-width=637&scale=1.0)
2g |
3g | 8 | 74% |
| 8 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-173-i15.svg?max-width=637&scale=1.0)
2h |
3h | 14 | 86% |
| 9 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-173-i16.svg?max-width=637&scale=1.0)
2i |
3i | 20 | 80% |
| 10 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-173-i17.svg?max-width=637&scale=1.0)
2j |
3j | 13 | 86% |
| 11 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-173-i18.svg?max-width=637&scale=1.0)
2k |
3k | 15 | 66% |
| 12 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-173-i19.svg?max-width=637&scale=1.0)
2l |
3l | 15 | 64% |
| 13 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-173-i20.svg?max-width=637&scale=1.0)
2m |
3m | 31 | 62% |
| 14 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-173-i21.svg?max-width=637&scale=1.0)
2n |
3n | 30 | 65% |
aConditions: 2.6 mmol of arylboronic acid 2a–n, 1.6 mmol of 1a, 10 mol % of a catalyst, 1.6 mmol of NaOH, methanol, reflux, air bubbled through the solution. The progress of the reaction was monitored by TLC. bIsolated yields.
The yields also vary with the location of the substituent on the benzene ring. Ortho- and meta-substituted phenylboronic acids gave in most cases lower yields in comparison to para-substituted ones (Table 5, entries 2,3,5–8,11–14). This might be a result of steric hindrance around the reaction center in the catalytic cycle.
Applicability of the synthetic procedure to the preparation of various N-aryl-C-nitroazoles was also investigated. For this purpose the cross coupling of phenylboronic acid with a series of C-nitro-NH-azoles such as: 3(5)-nitropyrazole (1a) (pKa = 9.81) [46], 4-nitropyrazole (1b) (pKa = 9.67) [46], 4(5)-nitroimidazole (1c) (pKa = 8.93) [47], 3-nitro-1,2,4-triazole (1d) (pKa = 6.05) [48] and 2-nitroimidazole (1e) (pKa = 7.15 in CH3OH/H2O 1:1) [49] (Scheme 4, Table 6) was carried out. The results show that this method allows N-aryl derivatives of C-nitroazoles to be obtained within a wide pKa range.
![[1860-5397-9-173-i4]](/bjoc/content/inline/1860-5397-9-173-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cross coupling of C-nitro-NH-azoles 1a–d with phenylboronic acid (2a).
Scheme 4: Cross coupling of C-nitro-NH-azoles 1a–d with phenylboronic acid (2a).
Table 6: Cross coupling of C-nitro-NH-azoles 1a–e with 2a.a
| Entry | C-nitro-NH-azole | Product | Time [h] | Yieldb |
|---|---|---|---|---|
| 1 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-173-i22.svg?max-width=637&scale=1.0)
1a |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-173-i23.svg?max-width=637&scale=1.0)
3a |
18 | 82% |
| 2 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-173-i24.svg?max-width=637&scale=1.0)
1b |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-173-i25.svg?max-width=637&scale=1.0)
3o |
15 | 53% |
| 3 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-173-i26.svg?max-width=637&scale=1.0)
1c |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-173-i27.svg?max-width=637&scale=1.0)
3p |
18 | 86% |
| 4 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-173-i28.svg?max-width=637&scale=1.0)
1d |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-173-i29.svg?max-width=637&scale=1.0)
3r |
16 | 50% |
| 5 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-173-i30.svg?max-width=637&scale=1.0)
1e |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-173-i31.svg?max-width=637&scale=1.0)
3s |
9 | 40% |
aConditions: 2.6 mmol of 2a, 1.6 mmol of C-nitroazole 1a–e, 10 mol % of a catalyst, 1.6 mmol of NaOH, methanol, reflux, air bubbled through the solution. The progress of the reaction was controlled by TLC. bIsolated yields.
Comparing our results presented here with those reported very recently by V. A. Chertkov et al. [20], it appears that both methods are regioselective and lead to syntheses of N-aryl-C-nitroazoles in moderate to high yields. Arylboronic acids are more easily available than diaryliodonium salts, and thus the scope of our approach seems to be wider. The much milder reaction conditions presented in this paper make this route more valuable.
Conclusion
In conclusion, we have developed an efficient and simple method for the cross coupling of arylboronic acids with C-nitro-NH-azoles in the presence of a catalytic amount of simple copper salts. The reaction takes place in a protic solvent containing a base, both of which are necessary for providing good yields of the products. The method represents an important supplement to the synthetic methodologies for the preparation of N-aryl-C-nitroazoles and can be successfully applied to the synthesis of a series of diverse C-nitroazoles functionalized with an aryl substituent on a ring nitrogen atom.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization of products. | ||
| Format: PDF | Size: 354.9 KB | Download |
| Supporting Information File 2: Crystallographic information file (structure of 3-nitro-1-[4-(trifluoromethoxy)phenyl]-3-nitro-1H-pyrazole). | ||
| Format: CIF | Size: 15.8 KB | Download |
References
-
Wang, S.; Sun, H.; Nikolovsksa-Colska, Z.; Yang, C.-Y.; Xu, L.; Saito, N. G.; Chen, J. Conformationally constrained smac mimetics and the uses thereof. U.S. Patent 7,674,787, March 9, 2010.
Return to citation in text: [1] -
Dereu, N.; Welter, A.; Ghyczy, M. Herbicidally active 1-substituted 2,4(5)-diisopropyl 5(4)-nitroimidazoles. German Patent 3,247,648, June 28, 1984.
Return to citation in text: [1] -
Girard, M.; Clairmont, F.; Maneckjee, A.; Mousseau, N.; Dawson, B. A.; Whitehouse, L. W. Can. J. Chem. 1993, 71, 1349–1352. doi:10.1139/v93-174
Return to citation in text: [1] -
Spear, R. J. Aust. J. Chem. 1984, 37, 2453–2468. doi:10.1071/CH9842453
Return to citation in text: [1] -
Grimmet, M. R. Imidazoles and their Benzo Derivatives: (iii) Synthesis and Applications. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon: Oxford, 1984; Vol. 5, pp 469–498.
Return to citation in text: [1] -
Potts, K. T. Chem. Rev. 1961, 61, 87–127. doi:10.1021/cr60210a001
Return to citation in text: [1] -
Davis, D. P.; Kirk, K. L.; Cohen, L. A. J. Heterocycl. Chem. 1982, 19, 253–256. doi:10.1002/jhet.5570190206
Return to citation in text: [1] -
Makarova, N. G.; Anisimova, N. A.; Berkova, G. A.; Deiko, L. I.; Berestovitskaya, V. M. Russ. J. Org. Chem. 2005, 41, 941–943. doi:10.1007/s11178-005-0272-1
Return to citation in text: [1] -
Larina, L.; Lopyrev, V. Application of Nitroazoles. Nitroazoles: Synthesis, Structure and Applications; Topics in Applied Chemistry; Springer Verlag: New York, NY, USA, 2009; pp 407–432. doi:10.1007/978-0-387-98070-6_4
And references cited therein.
Return to citation in text: [1] -
Bourdin Trunz, B.; Jędrysiak, R.; Tweats, D.; Brun, R.; Kaiser, M.; Suwiński, J.; Torreele, E. Eur. J. Med. Chem. 2011, 46, 1524–1535. doi:10.1016/j.ejmech.2011.01.071
Return to citation in text: [1] -
Walczak, K.; Gondela, A.; Suwiński, J. Eur. J. Med. Chem. 2004, 39, 849–853. doi:10.1016/j.ejmech.2004.06.014
Return to citation in text: [1] -
Kurpet, M.; Jędrysiak, R.; Suwiński, J. Chem. Heterocycl. Compd. 2013, 48, 1737–1750. doi:10.1007/s10593-013-1205-5
Return to citation in text: [1] -
Grimmett, M. R.; Hartshorn, S. R.; Schofield, K.; Weston, J. B. J. Chem. Soc., Perkin Trans. 2 1972, 1654–1660. doi:10.1039/P29720001654
Return to citation in text: [1] -
Pocar, D.; Maiorana, S.; Dalla Croce, P. Gazz. Chim. Ital. 1968, 98, 949–957.
Return to citation in text: [1] -
Nishiwaki, N.; Ogihara, T.; Takami, T.; Tamura, M.; Ariga, M. J. Org. Chem. 2004, 69, 8382–8386. doi:10.1021/jo0488513
Return to citation in text: [1] -
Jędrysiak, R.; Sawicki, M.; Wagner, P.; Suwiński, J. ARKIVOC 2007, vi, 103–111.
Return to citation in text: [1] -
Predvoditeleva, G. S.; Kartseva, T. V.; Shchukina, M. N. Pharm. Chem. J. 1974, 8, 525–527. doi:10.1007/BF00760681
Return to citation in text: [1] [2] [3] -
Coburn, M. D. J. Heterocycl. Chem. 1970, 7, 455–456. doi:10.1002/jhet.5570070242
Return to citation in text: [1] [2] [3] -
Chauzov, V. A.; Parchinsky, V. Z.; Sinelshchikova, E. V.; Petrosyan, V. A. Russ. Chem. Bull. 2001, 50, 1274–1279. doi:10.1023/A:1014023310543
Return to citation in text: [1] -
Chertkov, V. A.; Shestakova, A. K.; Davydov, D. V. Khim. Geterotsikl. Soedin. 2011, 1, 63–74.
Chem. Heterocycl. Compd. 2001, 47, 45–54. doi:10.1007/s10593-011-0718-z
Return to citation in text: [1] [2] [3] [4] [5] -
Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winteres, M. P. Tetrahedron Lett. 1998, 39, 2933–2936. doi:10.1016/S0040-4039(98)00503-6
Return to citation in text: [1] [2] [3] -
Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/S0040-4039(98)00504-8
Return to citation in text: [1] [2] [3] [4] -
Collman, J. P.; Zhong, M. Org. Lett. 2000, 2, 1233–1236. doi:10.1021/ol000033j
Return to citation in text: [1] [2] [3] -
Lan, J.-B.; Chen, L.; Yu, X.-Q.; You, J.-S.; Xie, R.-G. Chem. Commun. 2004, 188–189. doi:10.1039/b307734a
Return to citation in text: [1] [2] [3] -
Kantam, M. L.; Venkanna, G. T.; Sridhar, C.; Sreedhar, B.; Choudary, B. M. J. Org. Chem. 2006, 71, 9522–9524. doi:10.1021/jo0614036
Return to citation in text: [1] [2] -
Chen, S.; Huang, H.; Liu, X.; Shen, J.; Jiang, H.; Liu, H. J. Comb. Chem. 2008, 10, 358–360. doi:10.1021/cc8000053
Return to citation in text: [1] -
Reddy, K. R.; Kumar, N. S.; Sreedhar, B.; Kantam, M. L. J. Mol. Catal. A 2006, 252, 136–141. doi:10.1016/j.molcata.2006.02.053
Return to citation in text: [1] -
Filipski, K. J.; Bian, J.; Ebner, D. C.; Lee, E. C. Y.; Li, J.-C.; Sammons, M. F.; Wright, S. W.; Stevens, B. D.; Didiuk, M. T.; Tu, M.; Perreault, C.; Brown, J.; Atkinson, K.; Tan, B.; Salatto, C. T.; Litchfield, J.; Pfefferkorn, J. A.; Guzman-Perez, A. Bioorg. Med. Chem. Lett. 2012, 22, 415–420. doi:10.1016/j.bmcl.2011.10.113
Return to citation in text: [1] [2] -
Berk, S. C.; Close, J.; Hamblett, C.; Heidebrecht, R. W.; Kattar, S. D.; Kliman, L. T.; Mampreian, D. M.; Methot, J. L.; Miller, T.; Sloman, D. L.; Stanton, M. G.; Tempest, P.; Zabierek, A. A. Spirocyclic compounds. U.S. Patent 7,544,695, June 9, 2009.
Return to citation in text: [1] -
Goergens, U.; Mihara, J.; Murata, T.; Shibuya, K.; Shimojo, E.; Yamazaki, D.; Yoneta, Y. Insecticidal aryl pyrrolidines. WO Patent Application 2008/128711 A1, Oct 30, 2008.
Return to citation in text: [1] -
Choi, H.-S.; Ellis, A. D.; He, X.; Liu, H.; Nguyen, N. T.; Wang, Z.; Woodmansee, D.; Wu, B.; Yang, K. Compounds and compositions as inhibitors of cannabinoid receptor 1 activity. WO Patent Application 2006/047516 A2, May 4, 2006.
Return to citation in text: [1] -
Lewis, J. G.; Jacobs, J. W.; Reich, N.; Leadbetter, R.; Bell, N.; Chang, H.-T.; Chen, T.; Navre, M.; Charmot, D.; Carreras, Ch.; Labonte, E. Compounds and methods for inhibiting phosphate transport. WO Patent Application 2012/006473 A1, Jan 12, 2012.
Return to citation in text: [1] -
Dounay, A. B.; Mcallister, L. A.; Parikh, V. D.; Rong, S.; Verhoest, P. R. Kat II inhibitors. WO Patent Application 2012/073143 A1, June 17, 2012.
Return to citation in text: [1] -
Grimmett, M. R.; Lim, K. H. R.; Weavers, R. T. Aust. J. Chem. 1979, 32, 2203–2213. doi:10.1071/CH9792203
Return to citation in text: [1] -
Larina, L.; Lopyrev, V. Application of Nitroazoles. Nitroazoles: Synthesis, Structure and Applications; Topics in Applied Chemistry; Springer: New York, NY, USA, 2009; pp 182–199. doi:10.1007/978-0-387-98070-6
And references cited therein.
Return to citation in text: [1] [2] -
Collman, J. P.; Zhong, M.; Zhang, C.; Costanzo, S. J. Org. Chem. 2001, 66, 7892–7897. doi:10.1021/jo010615u
Return to citation in text: [1] [2] [3] -
Kantam, M. L.; Prakash, B. V.; Reddy, Ch. V. J. Mol. Catal. A 2005, 241, 162–165. doi:10.1016/j.molcata.2005.07.015
Return to citation in text: [1] [2] -
Evans, D. A.; Katz, J. L.; West, T. R. Tetrahedron Lett. 1998, 39, 2937–2940. doi:10.1016/S0040-4039(98)00502-4
Return to citation in text: [1] -
Collman, J. P.; Zhong, M.; Zeng, L.; Costanzo, S. J. Org. Chem. 2001, 66, 1528–1531. doi:10.1021/jo0016780
Return to citation in text: [1] -
van Berkel, S. S.; van den Hoogenband, A.; Terpstra, J. W.; Tromp, M.; van Leeuwen, P. W. N. M.; van Strijdonck, G. P. F. Tetrahedron Lett. 2004, 45, 7659–7662. doi:10.1016/j.tetlet.2004.08.094
Return to citation in text: [1] -
Smith, M. B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007; p 362.
Return to citation in text: [1] -
Faltin, C.; Fleming, E. M.; Connon, S. J. J. Org. Chem. 2004, 69, 6496–6499. doi:10.1021/jo0490907
Return to citation in text: [1] -
Reeves, R. R. Recovery of alcohols. U.S. Patent 4,594,466, June 10, 1986.
Return to citation in text: [1] -
Granitza, D.; Beyermann, M.; Wenschuh, H.; Haber, H.; Carpino, L. A.; Truran, G. A.; Bienert, M. J. Chem. Soc., Chem. Commun. 1995, 2223–2224.
Return to citation in text: [1] -
Boncel, S.; Saletra, K.; Hefczyc, B.; Walczak, K. Z. Beilstein J. Org. Chem. 2011, 7, 173–178. doi:10.3762/bjoc.7.24
Return to citation in text: [1] -
Kanishchev, M. I.; Korneeva, N. V.; Shevelev, S. A.; Fainzil'berg, A. A. Chem. Heterocycl. Compd. 1988, 24, 353–370. doi:10.1007/BF00478852
Return to citation in text: [1] [2] -
Li, W.; Norris, B. C.; Snodgrass, P.; Prasad, K.; Stockett, A. S.; Pryamitsyn, V.; Ganesan, V.; Bielawski, C. W.; Manthiram, A. J. Phys. Chem. B 2009, 113, 10063–10067. doi:10.1021/jp904192t
Return to citation in text: [1] -
Kofman, T. P.; Kartseva, G. Yu. Russ. J. Org. Chem. 2001, 37, 707–716. doi:10.1023/A:1012460103654
Return to citation in text: [1] -
Gallo, G. G.; Pasqualucci, C. R.; Radaelli, P.; Lancini, G. C. J. Org. Chem. 1964, 29, 862–865. doi:10.1021/jo01027a023
Return to citation in text: [1]
| 20. |
Chertkov, V. A.; Shestakova, A. K.; Davydov, D. V. Khim. Geterotsikl. Soedin. 2011, 1, 63–74.
Chem. Heterocycl. Compd. 2001, 47, 45–54. doi:10.1007/s10593-011-0718-z |
| 34. | Grimmett, M. R.; Lim, K. H. R.; Weavers, R. T. Aust. J. Chem. 1979, 32, 2203–2213. doi:10.1071/CH9792203 |
| 17. | Predvoditeleva, G. S.; Kartseva, T. V.; Shchukina, M. N. Pharm. Chem. J. 1974, 8, 525–527. doi:10.1007/BF00760681 |
| 22. | Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/S0040-4039(98)00504-8 |
| 24. | Lan, J.-B.; Chen, L.; Yu, X.-Q.; You, J.-S.; Xie, R.-G. Chem. Commun. 2004, 188–189. doi:10.1039/b307734a |
| 36. | Collman, J. P.; Zhong, M.; Zhang, C.; Costanzo, S. J. Org. Chem. 2001, 66, 7892–7897. doi:10.1021/jo010615u |
| 37. | Kantam, M. L.; Prakash, B. V.; Reddy, Ch. V. J. Mol. Catal. A 2005, 241, 162–165. doi:10.1016/j.molcata.2005.07.015 |
| 38. | Evans, D. A.; Katz, J. L.; West, T. R. Tetrahedron Lett. 1998, 39, 2937–2940. doi:10.1016/S0040-4039(98)00502-4 |
| 35. |
Larina, L.; Lopyrev, V. Application of Nitroazoles. Nitroazoles: Synthesis, Structure and Applications; Topics in Applied Chemistry; Springer: New York, NY, USA, 2009; pp 182–199. doi:10.1007/978-0-387-98070-6
And references cited therein. |
| 24. | Lan, J.-B.; Chen, L.; Yu, X.-Q.; You, J.-S.; Xie, R.-G. Chem. Commun. 2004, 188–189. doi:10.1039/b307734a |
| 17. | Predvoditeleva, G. S.; Kartseva, T. V.; Shchukina, M. N. Pharm. Chem. J. 1974, 8, 525–527. doi:10.1007/BF00760681 |
| 35. |
Larina, L.; Lopyrev, V. Application of Nitroazoles. Nitroazoles: Synthesis, Structure and Applications; Topics in Applied Chemistry; Springer: New York, NY, USA, 2009; pp 182–199. doi:10.1007/978-0-387-98070-6
And references cited therein. |
| 20. |
Chertkov, V. A.; Shestakova, A. K.; Davydov, D. V. Khim. Geterotsikl. Soedin. 2011, 1, 63–74.
Chem. Heterocycl. Compd. 2001, 47, 45–54. doi:10.1007/s10593-011-0718-z |
| 18. | Coburn, M. D. J. Heterocycl. Chem. 1970, 7, 455–456. doi:10.1002/jhet.5570070242 |
| 39. | Collman, J. P.; Zhong, M.; Zeng, L.; Costanzo, S. J. Org. Chem. 2001, 66, 1528–1531. doi:10.1021/jo0016780 |
| 40. | van Berkel, S. S.; van den Hoogenband, A.; Terpstra, J. W.; Tromp, M.; van Leeuwen, P. W. N. M.; van Strijdonck, G. P. F. Tetrahedron Lett. 2004, 45, 7659–7662. doi:10.1016/j.tetlet.2004.08.094 |
| 41. | Smith, M. B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007; p 362. |
| 23. | Collman, J. P.; Zhong, M. Org. Lett. 2000, 2, 1233–1236. doi:10.1021/ol000033j |
| 36. | Collman, J. P.; Zhong, M.; Zhang, C.; Costanzo, S. J. Org. Chem. 2001, 66, 7892–7897. doi:10.1021/jo010615u |
| 25. | Kantam, M. L.; Venkanna, G. T.; Sridhar, C.; Sreedhar, B.; Choudary, B. M. J. Org. Chem. 2006, 71, 9522–9524. doi:10.1021/jo0614036 |
| 37. | Kantam, M. L.; Prakash, B. V.; Reddy, Ch. V. J. Mol. Catal. A 2005, 241, 162–165. doi:10.1016/j.molcata.2005.07.015 |
| 45. | Boncel, S.; Saletra, K.; Hefczyc, B.; Walczak, K. Z. Beilstein J. Org. Chem. 2011, 7, 173–178. doi:10.3762/bjoc.7.24 |
| 21. | Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winteres, M. P. Tetrahedron Lett. 1998, 39, 2933–2936. doi:10.1016/S0040-4039(98)00503-6 |
| 22. | Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/S0040-4039(98)00504-8 |
| 44. | Granitza, D.; Beyermann, M.; Wenschuh, H.; Haber, H.; Carpino, L. A.; Truran, G. A.; Bienert, M. J. Chem. Soc., Chem. Commun. 1995, 2223–2224. |
| 42. | Faltin, C.; Fleming, E. M.; Connon, S. J. J. Org. Chem. 2004, 69, 6496–6499. doi:10.1021/jo0490907 |
| 21. | Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winteres, M. P. Tetrahedron Lett. 1998, 39, 2933–2936. doi:10.1016/S0040-4039(98)00503-6 |
| 22. | Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/S0040-4039(98)00504-8 |
| 23. | Collman, J. P.; Zhong, M. Org. Lett. 2000, 2, 1233–1236. doi:10.1021/ol000033j |
| 36. | Collman, J. P.; Zhong, M.; Zhang, C.; Costanzo, S. J. Org. Chem. 2001, 66, 7892–7897. doi:10.1021/jo010615u |
| 46. | Kanishchev, M. I.; Korneeva, N. V.; Shevelev, S. A.; Fainzil'berg, A. A. Chem. Heterocycl. Compd. 1988, 24, 353–370. doi:10.1007/BF00478852 |
| 47. | Li, W.; Norris, B. C.; Snodgrass, P.; Prasad, K.; Stockett, A. S.; Pryamitsyn, V.; Ganesan, V.; Bielawski, C. W.; Manthiram, A. J. Phys. Chem. B 2009, 113, 10063–10067. doi:10.1021/jp904192t |
| 46. | Kanishchev, M. I.; Korneeva, N. V.; Shevelev, S. A.; Fainzil'berg, A. A. Chem. Heterocycl. Compd. 1988, 24, 353–370. doi:10.1007/BF00478852 |
| 1. | Wang, S.; Sun, H.; Nikolovsksa-Colska, Z.; Yang, C.-Y.; Xu, L.; Saito, N. G.; Chen, J. Conformationally constrained smac mimetics and the uses thereof. U.S. Patent 7,674,787, March 9, 2010. |
| 5. | Grimmet, M. R. Imidazoles and their Benzo Derivatives: (iii) Synthesis and Applications. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon: Oxford, 1984; Vol. 5, pp 469–498. |
| 6. | Potts, K. T. Chem. Rev. 1961, 61, 87–127. doi:10.1021/cr60210a001 |
| 17. | Predvoditeleva, G. S.; Kartseva, T. V.; Shchukina, M. N. Pharm. Chem. J. 1974, 8, 525–527. doi:10.1007/BF00760681 |
| 18. | Coburn, M. D. J. Heterocycl. Chem. 1970, 7, 455–456. doi:10.1002/jhet.5570070242 |
| 3. | Girard, M.; Clairmont, F.; Maneckjee, A.; Mousseau, N.; Dawson, B. A.; Whitehouse, L. W. Can. J. Chem. 1993, 71, 1349–1352. doi:10.1139/v93-174 |
| 14. | Pocar, D.; Maiorana, S.; Dalla Croce, P. Gazz. Chim. Ital. 1968, 98, 949–957. |
| 15. | Nishiwaki, N.; Ogihara, T.; Takami, T.; Tamura, M.; Ariga, M. J. Org. Chem. 2004, 69, 8382–8386. doi:10.1021/jo0488513 |
| 2. | Dereu, N.; Welter, A.; Ghyczy, M. Herbicidally active 1-substituted 2,4(5)-diisopropyl 5(4)-nitroimidazoles. German Patent 3,247,648, June 28, 1984. |
| 16. | Jędrysiak, R.; Sawicki, M.; Wagner, P.; Suwiński, J. ARKIVOC 2007, vi, 103–111. |
| 10. | Bourdin Trunz, B.; Jędrysiak, R.; Tweats, D.; Brun, R.; Kaiser, M.; Suwiński, J.; Torreele, E. Eur. J. Med. Chem. 2011, 46, 1524–1535. doi:10.1016/j.ejmech.2011.01.071 |
| 12. | Kurpet, M.; Jędrysiak, R.; Suwiński, J. Chem. Heterocycl. Compd. 2013, 48, 1737–1750. doi:10.1007/s10593-013-1205-5 |
| 20. |
Chertkov, V. A.; Shestakova, A. K.; Davydov, D. V. Khim. Geterotsikl. Soedin. 2011, 1, 63–74.
Chem. Heterocycl. Compd. 2001, 47, 45–54. doi:10.1007/s10593-011-0718-z |
| 9. |
Larina, L.; Lopyrev, V. Application of Nitroazoles. Nitroazoles: Synthesis, Structure and Applications; Topics in Applied Chemistry; Springer Verlag: New York, NY, USA, 2009; pp 407–432. doi:10.1007/978-0-387-98070-6_4
And references cited therein. |
| 13. | Grimmett, M. R.; Hartshorn, S. R.; Schofield, K.; Weston, J. B. J. Chem. Soc., Perkin Trans. 2 1972, 1654–1660. doi:10.1039/P29720001654 |
| 8. | Makarova, N. G.; Anisimova, N. A.; Berkova, G. A.; Deiko, L. I.; Berestovitskaya, V. M. Russ. J. Org. Chem. 2005, 41, 941–943. doi:10.1007/s11178-005-0272-1 |
| 48. | Kofman, T. P.; Kartseva, G. Yu. Russ. J. Org. Chem. 2001, 37, 707–716. doi:10.1023/A:1012460103654 |
| 7. | Davis, D. P.; Kirk, K. L.; Cohen, L. A. J. Heterocycl. Chem. 1982, 19, 253–256. doi:10.1002/jhet.5570190206 |
| 11. | Walczak, K.; Gondela, A.; Suwiński, J. Eur. J. Med. Chem. 2004, 39, 849–853. doi:10.1016/j.ejmech.2004.06.014 |
| 49. | Gallo, G. G.; Pasqualucci, C. R.; Radaelli, P.; Lancini, G. C. J. Org. Chem. 1964, 29, 862–865. doi:10.1021/jo01027a023 |
| 23. | Collman, J. P.; Zhong, M. Org. Lett. 2000, 2, 1233–1236. doi:10.1021/ol000033j |
| 24. | Lan, J.-B.; Chen, L.; Yu, X.-Q.; You, J.-S.; Xie, R.-G. Chem. Commun. 2004, 188–189. doi:10.1039/b307734a |
| 25. | Kantam, M. L.; Venkanna, G. T.; Sridhar, C.; Sreedhar, B.; Choudary, B. M. J. Org. Chem. 2006, 71, 9522–9524. doi:10.1021/jo0614036 |
| 26. | Chen, S.; Huang, H.; Liu, X.; Shen, J.; Jiang, H.; Liu, H. J. Comb. Chem. 2008, 10, 358–360. doi:10.1021/cc8000053 |
| 27. | Reddy, K. R.; Kumar, N. S.; Sreedhar, B.; Kantam, M. L. J. Mol. Catal. A 2006, 252, 136–141. doi:10.1016/j.molcata.2006.02.053 |
| 19. | Chauzov, V. A.; Parchinsky, V. Z.; Sinelshchikova, E. V.; Petrosyan, V. A. Russ. Chem. Bull. 2001, 50, 1274–1279. doi:10.1023/A:1014023310543 |
| 20. |
Chertkov, V. A.; Shestakova, A. K.; Davydov, D. V. Khim. Geterotsikl. Soedin. 2011, 1, 63–74.
Chem. Heterocycl. Compd. 2001, 47, 45–54. doi:10.1007/s10593-011-0718-z |
| 21. | Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winteres, M. P. Tetrahedron Lett. 1998, 39, 2933–2936. doi:10.1016/S0040-4039(98)00503-6 |
| 22. | Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941–2944. doi:10.1016/S0040-4039(98)00504-8 |
| 33. | Dounay, A. B.; Mcallister, L. A.; Parikh, V. D.; Rong, S.; Verhoest, P. R. Kat II inhibitors. WO Patent Application 2012/073143 A1, June 17, 2012. |
| 18. | Coburn, M. D. J. Heterocycl. Chem. 1970, 7, 455–456. doi:10.1002/jhet.5570070242 |
| 31. | Choi, H.-S.; Ellis, A. D.; He, X.; Liu, H.; Nguyen, N. T.; Wang, Z.; Woodmansee, D.; Wu, B.; Yang, K. Compounds and compositions as inhibitors of cannabinoid receptor 1 activity. WO Patent Application 2006/047516 A2, May 4, 2006. |
| 32. | Lewis, J. G.; Jacobs, J. W.; Reich, N.; Leadbetter, R.; Bell, N.; Chang, H.-T.; Chen, T.; Navre, M.; Charmot, D.; Carreras, Ch.; Labonte, E. Compounds and methods for inhibiting phosphate transport. WO Patent Application 2012/006473 A1, Jan 12, 2012. |
| 28. | Filipski, K. J.; Bian, J.; Ebner, D. C.; Lee, E. C. Y.; Li, J.-C.; Sammons, M. F.; Wright, S. W.; Stevens, B. D.; Didiuk, M. T.; Tu, M.; Perreault, C.; Brown, J.; Atkinson, K.; Tan, B.; Salatto, C. T.; Litchfield, J.; Pfefferkorn, J. A.; Guzman-Perez, A. Bioorg. Med. Chem. Lett. 2012, 22, 415–420. doi:10.1016/j.bmcl.2011.10.113 |
| 30. | Goergens, U.; Mihara, J.; Murata, T.; Shibuya, K.; Shimojo, E.; Yamazaki, D.; Yoneta, Y. Insecticidal aryl pyrrolidines. WO Patent Application 2008/128711 A1, Oct 30, 2008. |
| 20. |
Chertkov, V. A.; Shestakova, A. K.; Davydov, D. V. Khim. Geterotsikl. Soedin. 2011, 1, 63–74.
Chem. Heterocycl. Compd. 2001, 47, 45–54. doi:10.1007/s10593-011-0718-z |
| 28. | Filipski, K. J.; Bian, J.; Ebner, D. C.; Lee, E. C. Y.; Li, J.-C.; Sammons, M. F.; Wright, S. W.; Stevens, B. D.; Didiuk, M. T.; Tu, M.; Perreault, C.; Brown, J.; Atkinson, K.; Tan, B.; Salatto, C. T.; Litchfield, J.; Pfefferkorn, J. A.; Guzman-Perez, A. Bioorg. Med. Chem. Lett. 2012, 22, 415–420. doi:10.1016/j.bmcl.2011.10.113 |
| 29. | Berk, S. C.; Close, J.; Hamblett, C.; Heidebrecht, R. W.; Kattar, S. D.; Kliman, L. T.; Mampreian, D. M.; Methot, J. L.; Miller, T.; Sloman, D. L.; Stanton, M. G.; Tempest, P.; Zabierek, A. A. Spirocyclic compounds. U.S. Patent 7,544,695, June 9, 2009. |
© 2013 Kurpet et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)