Abstract
Carboxylic acids have been electro-oxidatively coupled to electron-rich olefins to form lactones. Kolbe decarboxylation does not appear to be a significant competing pathway. Experimental results indicate that oxidation occurs at the olefin and that the reaction proceeds through a radical cation intermediate.

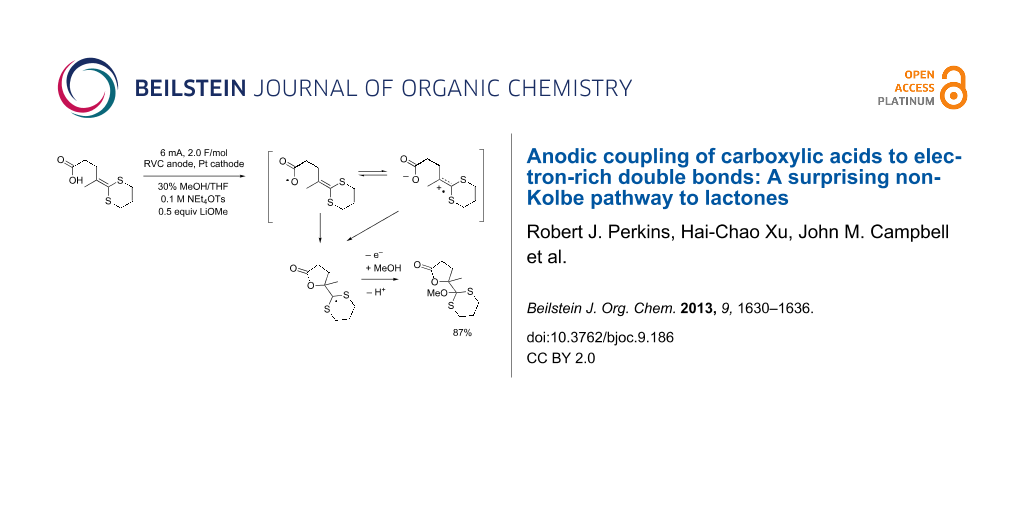
Graphical Abstract
Introduction
Anodic cyclization reactions of the general type shown in Scheme 1 can provide a powerful method for the synthesis of a variety of ring systems [1,2]. The reactions effectively reverse the polarity of an electron-rich functional group and in so doing open up entirely new opportunities for bond formation. In addition, the oxidative reactions lead to products that either preserve or increase the level of functionality found in the initial substrate. This provides synthetic handles for further development of the cyclic products generated.
![[1860-5397-9-186-i1]](/bjoc/content/inline/1860-5397-9-186-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General scheme for anodic cyclization reactions.
Scheme 1: General scheme for anodic cyclization reactions.
Such reactions can be used to build a variety of fused, bridged, and spirocyclic carbocyclic systems [3], cyclic amino acid derivatives [4], cyclic ethers [5,6], and lactones [7,8]. In most of these examples, the reactions can be viewed as arising from an oxidation that forms an olefinic radical cation that is then rapidly trapped by a nucleophile. This triggers a cascade of reactions that typically involve the loss of two protons (or a silyl group), a second oxidation of a radical intermediate, and solvent trapping. This reaction cascade leads to formation of the final product.
A combination of computational studies and competition experiments has begun to identify both cyclization reactions that proceed through different mechanistic pathways and cyclization reactions where the cascade of reactions downstream from the cyclization plays an important role in product formation [9]. For example, consider the competition experiment highlighted in Scheme 2. In this experiment, an electron-rich olefin was coupled to one of two competing nucleophiles, a sulfonamide and an alcohol (5). When the oxidation was run with 2,6-lutidine as a base (not shown), the reaction led to the formation of a radical cation from the electron-rich olefin followed by trapping by the alcohol nucleophile to form product 9. No product from sulfonamide trapping (8) was observed. However, when the reaction was run under more basic conditions (0.5 equivalents of lithium methoxide) cyclic voltammetry data indicated that the oxidative cyclizations were initiated by the oxidation of sulfonamide anion. This led to initial formation of N-localized radical 6. Depending on the reaction conditions, the experiment favoured either a sulfonamide or alcohol based cyclization. Density functional theory (DFT) calculations suggested that sulfonamide based cyclizations proceed by addition of the N-localized radical to the electron-rich double bond, a reaction that led to product 8. In a competitive process, an intramolecular electron-transfer reaction occurred that led to formation of a radical cation from the olefin 7 and regeneration of the sulfonamide anion. Formation of a radical cation from the olefin led to the possibility of alcohol trapping and the formation of cyclic ether product 9. Experimental data suggested that the alcohol trapping pathway provided the kinetic product but that the cyclization was reversible. The sulfonamide radical pathway afforded the thermodynamic product. In a very interesting development, we found that the reaction could be shifted toward the alcohol-trapping product by increasing the current passed through the cell. The increased current appeared to accelerate the removal of a second electron from an initial cyclic product like 3 (Scheme 1), a change that reduced the reversibility of the cyclization.
![[1860-5397-9-186-i2]](/bjoc/content/inline/1860-5397-9-186-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Anodic cyclization competition study.
Scheme 2: Anodic cyclization competition study.
Clearly, the use of basic reaction conditions with the acidic substrate led to dramatic mechanistic changes to our early, simplified view of the reactions. It is tempting to suggest that similar situations would arise with other acidic substrates. For example, would the oxidation of a substrate having a carboxylic acid and an electron-rich olefin lead to a carboxy radical pathway or a radical cation type reaction? To date, we have been hesitant to try such an experiment, and in fact we typically avoid substrates with carboxylic acid functional groups [5]. This action was taken because of the well-known Kolbe electrolysis reaction (Scheme 3) [10,11]. In the Kolbe electrolysis (Scheme 3, reaction 1), a carboxylic acid is oxidized. A decarboxylation reaction then leads to the formation of a radical that is subsequently trapped by a second radical formed in solution. The reaction has been used to form dimers [12], as well as in some cases cyclic products (Scheme 3, reaction 2) [13-15].
![[1860-5397-9-186-i3]](/bjoc/content/inline/1860-5397-9-186-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
However, the chemistry highlighted in Scheme 2 suggests that a Kolbe-type decarboxylation reaction might not interfere at all with an oxidative coupling reaction between a carboxylic acid and an electron-rich olefin. Examples in the literature of the coupling of carboxylic acids to aromatic rings, though limited in scope, were also encouraging [16-20].
There are three mechanistic possibilities which would allow for a successful reaction. First, if the carboxy radical 10 is formed, then it may simply add to the electron-rich olefin to give 12 faster than it undergoes a decarboxylation reaction (Scheme 4).
![[1860-5397-9-186-i4]](/bjoc/content/inline/1860-5397-9-186-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Oxidative coupling between a carboxylic acid and electron-rich olefin.
Scheme 4: Oxidative coupling between a carboxylic acid and electron-rich olefin.
Second, it is possible for a carboxy radical like 10 to undergo an intramolecular electron-transfer reaction analogous to that observed in the case of the sulfonamide-based radical. One might suspect the electron-transfer to form radical cation 11 to be even more favorable in the present case due to the stability of the resulting carboxylate anion. The electron-transfer would minimize the risk of decarboxylation and lead to carboxylate trapping of the radical cation to again form the cyclic radical 12.
A third mechanistic possibility is a direct oxidation of the elctron-rich olefin to form radical cation 11 followed by carboxylate trapping. All three mechanistic variations would then require a successful single-electron oxidation of 12 to ultimately lead to 13, the product of a direct oxidative coupling between the carboxylic acid moiety and the electron-rich olefin.
We report here that this is the case and that the direct oxidative coupling of a carboxylic acid and an electron-rich olefin can be accomplished in good yield. In all cases, the reactions appear to proceed through an olefinic radical cation intermediate.
Results and Discussion
Initial cyclization studies
The compatibility of the carboxylic acid with the anodic cyclization was initially studied by examining the anodic oxidation of substrates 14a–c (Table 1). The oxidation of 14a nicely afforded the five-membered ring product using either lithium methoxide or 2,6-lutidine as a base. Clearly, decarboxylation of the carboxylate anion was not a problem. In fact, cyclic voltammetry data suggest that the reaction originated from an oxidation of the ketene dithioacetal. The oxidation potential (Ep/2) for 14a was measured to be +0.68 V versus Ag/AgCl. For comparison, the oxidation potential measured for 10-undecenoic acid was +1.91 V versus Ag/AgCl in DMF solvent and +1.85 V versus Ag/AgCl in acetonitrile. When 0.5 equivalents of benzyltrimethylammonium hydroxide was added to the cyclic voltammetry solutions in order to generate the carboxylate and mimic the preparative oxidation conditions used for the reaction originating from 14a, the oxidation potential of the 10-undecenoic acid fell to 1.36–1.40 V versus Ag/AgCl for the DMF solution and 1.38 V versus Ag/AgCl for the acetonitrile solution. The carboxylate was only partially soluble in acetonitrile. While the formation of the carboxylate significantly lowered the oxidation potential of the acid moiety, it did not lower it close to the oxidation potential measured for substrate 14a or even the oxidation potential of a ketene dithioacetal (ca. +1.06 V versus Ag/AgCl) [21]. Hence, the oxidation potential measured for 14a is most consistent with a fast cyclization originating from oxidation of the olefin. Fast cyclization reactions are known to significantly lower the oxidation potential of a ketene dithioacetal. For example, the trapping of a ketene dithioacetal derived radical cation by an amine was shown to lower the oxidation potential measured for the ketene dithioacetal by 460 mV [21], a value consistent with the potential measured here for 14a.
Table 1: Anodic coupling of ketene dithioacetals and carboxylic acids.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-186-i6.svg?max-width=637&scale=1.0)
|
||||
| Substrate | n | Ep/2a | Base (0.5 equiv) | Yield (15) |
|---|---|---|---|---|
| 14a | 1 | 0.68 | LiOMe | 87% |
| 14a | 1 | 2,6-lutidine | 74% | |
| 14b | 2 | 0.71 | LiOMe | 0%b |
| 14b | 2 | 2,6-lutidine | 72% | |
| 14c | 3 | 1.06 | LiOMe | 0%c |
| 14c | 3 | 2,6-lutidine | 0% | |
aCyclic voltammetry data were obtained relative to a Ag/AgCl reference electrode with a sweep rate of 50 mV/s. b87% of the ring opened methyl ester 16b was obtained. c30% of the ring opened methyl ester 16c was obtained.
The oxidation of 14b to afford the six-membered ring product also proceeded well. When 2,6-lutidine was used as the base for the reaction, a 72% isolated yield of the desired product was obtained. In this case, the use of the more nucleophilic lithium methoxide as the base led to cyclization followed by opening of the lactone ring to form methyl ester product 16b. This product did have the olefin functionalized with an alcohol and a methoxy group indicating that the initial oxidative cyclization had occurred. In this case, the slightly higher oxidation potential measured for the substrate (+0.71 V versus Ag/AgCl) is consistent with a slightly slower cyclization reaction.
Attempts to generate seven-membered ring products using the oxidative cyclization were not successful. While 30% of a methoxide opened product 16c could be obtained with lithium methoxide as the base, no cyclic product was observed when 2,6-lutidine was employed. The reactions were quite messy and consistent with the formation of a radical cation followed by a cyclization that was too slow to compete with decomposition. The cyclic voltammetry data obtained were consistent with this observation. The +1.06 V versus Ag/AgCl oxidation potential measured for substrate 14c was the same as that measured for the ketene dithioacetal in the absence of a trapping group [21].
The anodic coupling of a carboxylic acid group with a vinyl sulfide and an enol ether were also examined (Table 2). As with earlier alcohol and amine based cyclizations, reactions with the vinyl sulfide coupling partner proceeded much better than did their enol ether counterparts [21,22]. In the previous cases, the argument was made that less polar radical cations underwent better trapping reactions with heteroatomic nucleophiles [23], and a similar argument can be made here.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-186-i7.svg?max-width=637&scale=1.0)
The oxidation potential for the vinyl sulfide used in substrate 17a is +1.08 V versus Ag/AgCl [9] and the oxidation potential of the enol ether in substrate 17b is +1.18 versus Ag/AgCl [9]. Both oxidation potentials are lower than the potential measured for the carboxylate suggesting that both reactions proceed through the olefinic radical cation. While an intramolecular electron-transfer to form the carboxy radical can occur, the difference in oxidation potentials suggests that the equilibrium would lie on the side of the oxidized olefin.
Styrene substrates and additional insights
With the ketene dithioacetal, vinyl sulfide, and enol ether substrates, it appeared clear that the initial oxidation was occurring at the olefin instead of at the carboxylate. We wondered how the reaction might change if the olefin had a higher oxidation potential. With this in mind, a series of carboxylic acid substituted styrene substrates were examined (Table 3).
Table 3: Extension to styrene derivatives.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-186-i8.svg?max-width=637&scale=1.0)
|
||||||
| Substrate | R | n | Ep/2a | Base/Temperature | F/mol | Yield |
|---|---|---|---|---|---|---|
| 19a | H | 1 | 1.52 | 0.5 equiv LiOMe/rt | 2 | 15% |
| 19a | H | 1 | 1.52 | 0.5 equiv LiOMe/rt | 10 | 27% |
| 19a | H | 1 | 1.52 | None/40 °C | 10 | 33% |
| 19b | H | 3 | 1.73 | NAb | NA | NA |
| 19c | 4-OMe | 1 | 1.31 | 0.5 equiv LiOMe/rt | 2 | 56% |
| 19c | 4-OMe | 1 | 1.31 | None/40 °C | 2 | 76% |
| 19d | 4-OMe | 3 | 1.42 | NA | NA | NA |
| 19e | 2-OMe | 1 | 1.39c | 0.5 equiv LiOMe/rt | 2 | 48% |
| 19e | 2-OMe | 1 | 1.39c | None/40 °C | 2 | 59% |
| 19f | 2-OMe | 3 | 1.39c | NA | NA | NA |
| 19g | 3-OMe | 1 | 1.42c | 0.5 equiv LiOMe/rt | 2 | 4% (NMR) |
| 19g | 3-OMe | 1 | 1.42c | 0.5 equiv LiOMe/rt | 10 | 35% |
| 19g | 3-OMe | 1 | 1.42c | None/40 °C | 10 | 23% |
| 19h | 3-OMe | 3 | 1.50 | NA | NA | NA |
| 19i | 2,4-OMe | 1 | 1.09 | 0.5 equiv LiOMe/rt | 2 | 48% |
| 19i | 2,4-OMe | 1 | 1.09 | None/40 °C | 2 | 45% |
| 19i | 2,4-OMe | 1 | 1.09 | 1.0 equiv LiOMe/rt | 2 | 74% |
| 19j | 2,4-OMe | 3 | 1.11 | NA | NA | NA |
aConditions: Substrates were dissolved in acetonitrile to a concentration of 0.025 M in a solution that contained 0.1 M tetraethylammonium tosylate. Cyclic voltammetry was performed at a sweep rate of 50 mV/s using a platinum anode. Half-wave oxidation potentials were measured versus a Ag/AgCl reference electrode. bNot applicable. cLithium perchlorate was used as the electrolyte.
The first substrate studied was the simple styrene derivative 19a (R = H). Cyclic voltammetry suggested that in the presence of base the initial oxidation should occur at the carboxylate (Ep/2 = +1.38 in acetonitrile) since the Ep/2 value measured for substrate 19a under neutral conditions was +1.52 V versus Ag/AgCl. Several sets of conditions were attempted to achieve the cyclization. However, in no case was the yield of cyclic product obtained more than 33%. Proton NMR data taken of the crude reaction mixture showed no evidence for decarboxylation. Instead, all products appeared to arise from a styrene derived radical cation, an intermediate that might be formed by an intramolecular electron-transfer. Furthermore, the recovery of starting material after 2 F/mol of current had been passed through the reaction mixture indicated poor current efficiency. Why might an increase in the oxidation potential of the olefin hurt the reaction?
Past experience suggests that in such cases the low yield of product obtained might arise from either a slow initial cyclization or the formation of a stable cyclic radical that then struggles to undergo the second oxidation. Both pathways might lead to the formation of a polymer or decomposition, and in so doing lower the mass balance for the reaction as observed. Also, diffusion of a stable radical or radical cation to the cathode would result in a reduction to give the starting material, a scenario that would lead to low current efficiency. Of the two mechanistic explanations for these observations, we believe that a slow second oxidation step is the problem.
The possibility of a slow cyclization was eliminated by consideration of the observed half-wave oxidation potential measured for 19a. The oxidation potential (Ep/2) of +1.52 V for 19a (measured under neutral conditions) was significantly lower than that measured for 19b (+1.73 V), a similar styrene with a longer tether between the coupling partners. This observation is consistent with the oxidation of 19a leading to a fast cyclization that rapidly removes the radical cation from the electrode surface, resulting in a lower observed oxidation potential [21]. The seven-membered ring cyclization arising from the oxidation of 19b would be significantly slower, giving rise to a higher oxidation potential.
Given the evidence for a fast cyclization reaction, we attributed the challenges associated with the styrene cyclizations to a problem with the second single-electron oxidation. This idea was supported by our next set of experiments. Methoxy-substituted styrenes 19c (R = 4-OMe), 19e (R = 2-OMe), and 19g (R = 3-OMe) were prepared. Substrate 19c has a p-methoxy substituent positioned perfectly for aiding in the removal of a second electron from cyclic radical 21a (Scheme 5). Indeed, the reactions of 19c proceeded better than the cyclizations originating from 19a, giving acceptable yields and complete consumption of the starting material after 2 F/mol of current had been passed. In this case, the reaction benefited strongly from using less basic reaction conditions and slightly elevated temperatures. When more basic reaction conditions were used, an overoxidized product derived from the elimination of the lactone ester to form a methoxy styrene from product 20c was obtained. The elevated temperatures are known to assist the intramolecular cyclization. The significantly lower oxidation potential measured for 19c relative to that measured for its seven-membered ring counterpart 19d was again consistent with a fast initial cyclization.
![[1860-5397-9-186-i5]](/bjoc/content/inline/1860-5397-9-186-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Predicted relative rates of single-electron oxidation based on resonance stabilization of the resulting cation.
Scheme 5: Predicted relative rates of single-electron oxidation based on resonance stabilization of the resul...
We believe that the primary advantage of the methoxy group is to facilitate the second electron oxidation, rather than merely stabilizing an olefinic radical cation. To demonstrate this, the position of the methoxy group on the ring was altered systematically (see Table 3). Cyclic radical 21b, arising from the reaction of m-methoxy substituted styrene 19g (Table 3), would not be expected to oxidize as readily as radical 21a. Indeed, the anodic oxidation of 19g gave low yields and poor current efficiency, mirroring the results obtained from the oxidation of unsubstituted styrene 19a. As with other substrates, the oxidation potential of 19g was lower than that of its seven-membered ring counterpart 19h, indicating a fast cyclization. Hence, the poor yield obtained from the oxidation of 19g was not due to a problem with the first step.
Good yields and efficient consumption of starting material were regained upon moving the methoxy group to the ortho position (19e). Interestingly, 19e showed no oxidation potential drop from the analogous long-chain substrate 19f. It is tempting to assign this to a slow cyclization, but we do not believe that this is necessarily the case. The relatively low oxidation potentials for 19e and 19f (as well as the potentials measured for 19i and 19j) may be due to an interaction between the o-methoxy substituent and the radical cation (Figure 1). Such a stabilizing interaction would be expected to lower the oxidation potential of the olefin.
![[1860-5397-9-186-1]](/bjoc/content/figures/1860-5397-9-186-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Radical cation stabilization by an o-methoxy substituent.
Figure 1: Radical cation stabilization by an o-methoxy substituent.
We also investigated the reactivity of the o,p-dimethoxy substituted styrene 19i towards the coupling reaction. This reaction also led to a good yield of product and efficient consumption of the starting material. Surprisingly, the optimized conditions for the oxidation of 19i were different from those optimized for the oxidation of either 19c or 19e, and the initial yields for the cyclization of 19i were not good. The cyclization originating from 19i benefited from the use of more base. It appears that the use of base reduced the formation of a quinone methide from the product. For example, when a full equivalent of base was used for the reaction, the desired product was obtained in 74% isolated yield.
It is clear that the success of the styrene derived coupling reaction depends strongly on the position of the methoxy group on the aromatic ring. Ortho and para substitution leads to a successful cyclization while meta substitution does not. This observation is best explained by the reaction being dependent upon the ease with which the cyclic benzyl radicals 21a–c are oxidized.
Conclusion
It was found that carboxylic acid nucleophiles can be coupled to electron-rich olefins to form lactone products in good yield without interference from competitive decarboxylation reactions. The reactions are consistent with carboxylate trapping of an olefin-derived radical cation intermediate and depend strongly on the ease with which the second oxidation step in the mechanism occurs. This is particularly true when styrene-based substrates are used. Finally, optimization of reaction pH required careful consideration of product stability.
Supporting Information
| Supporting Information File 1: Procedures for electrolysis and cyclic voltammetry experiments, characterization of electrolysis products, procedures for synthesis and characterization of electrolysis starting materials. | ||
| Format: PDF | Size: 7.4 MB | Download |
References
-
Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/b512308a
Return to citation in text: [1] -
Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843
Return to citation in text: [1] -
Moeller, K. D. Synlett 2009, 1208–1218. doi:10.1055/s-0028-1088126
Return to citation in text: [1] -
Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2010, 132, 2839–2844. doi:10.1021/ja910586v
Return to citation in text: [1] -
Sutterer, A.; Moeller, K. D. J. Am. Chem. Soc. 2000, 122, 5636–5637. doi:10.1021/ja001063k
Return to citation in text: [1] [2] -
Liu, B.; Duan, S.; Sutterer, A. C.; Moeller, K. D. J. Am. Chem. Soc. 2002, 124, 10101–10111. doi:10.1021/ja026739l
Return to citation in text: [1] -
Brandt, J. D.; Moeller, K. D. Org. Lett. 2005, 7, 3553–3556. doi:10.1021/ol051296m
Return to citation in text: [1] -
Xu, H.-C.; Brandt, J. D.; Moeller, K. D. Tetrahedron Lett. 2008, 49, 3868–3871. doi:10.1016/j.tetlet.2008.04.075
Return to citation in text: [1] -
Campbell, J. M.; Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2012, 134, 18338–18344. doi:10.1021/ja307046j
Return to citation in text: [1] [2] [3] -
Torii, S.; Tanaka, H. Carboxylic Acids. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; Marcel Dekker: New York, 2001; pp 499–543.
Return to citation in text: [1] -
Schäfer, H. J. Recent Contributions of Kolbe Electrolysis to Organic Synthesis. In Electrochemistry IV; Steckhan, E., Ed.; Topics in Current Chemistry, Vol. 152; Springer-Verlag: New York, 1990; pp 91–151.
Return to citation in text: [1] -
Schäfer, H. J. Chem. Phys. Lipids 1979, 24, 321–333. doi:10.1016/0009-3084(79)90117-8
Return to citation in text: [1] -
Lebreux, F.; Buzzo, F.; Markó, I. E. Synlett 2008, 2815–2820. doi:10.1055/s-0028-1083547
Return to citation in text: [1] -
Huhtasaari, M.; Schäfer, H. J.; Becking, L. Angew. Chem., Int. Ed. Engl. 1984, 23, 980–981. doi:10.1002/anie.198409801
Return to citation in text: [1] -
Matzeit, A.; Schäfer, H. J.; Amatore, C. Synthesis 1995, 1432–1444. doi:10.1055/s-1995-4112
Return to citation in text: [1] -
Scott, A. I.; Dodson, P. A.; McCapra, F.; Meyers, M. B. J. Am. Chem. Soc. 1963, 85, 3702–3704. doi:10.1021/ja00905a040
Return to citation in text: [1] -
Coutts, I. G. C.; Edwards, M.; Musto, D. R.; Richards, D. J. Tetrahedron Lett. 1980, 21, 5055–5056. doi:10.1016/S0040-4039(00)71131-2
Return to citation in text: [1] -
Coutts, I. G. C.; Culbert, N. J.; Edwards, M.; Hadfield, J. A.; Musto, D. R.; Pavlidis, V. H.; Richards, D. J. J. Chem. Soc., Perkin Trans. 1 1985, 1829–1836. doi:10.1039/P19850001829
Return to citation in text: [1] -
Thomas, H. G.; Schwager, H.-W. Tetrahedron Lett. 1984, 25, 4471–4474. doi:10.1016/S0040-4039(01)81469-6
Return to citation in text: [1] -
Deffieux, D.; Fabre, I.; Courseille, C.; Quideau, S. J. Org. Chem. 2002, 67, 4458–4465. doi:10.1021/jo020023r
Return to citation in text: [1] -
Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924
Return to citation in text: [1] [2] [3] [4] [5] -
Xu, G.; Moeller, K. D. Org. Lett. 2010, 12, 2590–2593. doi:10.1021/ol100800u
Return to citation in text: [1] -
Tang, F.; Moeller, K. D. Tetrahedron 2009, 65, 10863–10875. doi:10.1016/j.tet.2009.09.028
Return to citation in text: [1]
| 21. | Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924 |
| 9. | Campbell, J. M.; Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2012, 134, 18338–18344. doi:10.1021/ja307046j |
| 9. | Campbell, J. M.; Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2012, 134, 18338–18344. doi:10.1021/ja307046j |
| 1. | Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/b512308a |
| 2. | Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843 |
| 7. | Brandt, J. D.; Moeller, K. D. Org. Lett. 2005, 7, 3553–3556. doi:10.1021/ol051296m |
| 8. | Xu, H.-C.; Brandt, J. D.; Moeller, K. D. Tetrahedron Lett. 2008, 49, 3868–3871. doi:10.1016/j.tetlet.2008.04.075 |
| 21. | Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924 |
| 22. | Xu, G.; Moeller, K. D. Org. Lett. 2010, 12, 2590–2593. doi:10.1021/ol100800u |
| 5. | Sutterer, A.; Moeller, K. D. J. Am. Chem. Soc. 2000, 122, 5636–5637. doi:10.1021/ja001063k |
| 6. | Liu, B.; Duan, S.; Sutterer, A. C.; Moeller, K. D. J. Am. Chem. Soc. 2002, 124, 10101–10111. doi:10.1021/ja026739l |
| 23. | Tang, F.; Moeller, K. D. Tetrahedron 2009, 65, 10863–10875. doi:10.1016/j.tet.2009.09.028 |
| 4. | Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2010, 132, 2839–2844. doi:10.1021/ja910586v |
| 21. | Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924 |
| 21. | Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924 |
| 12. | Schäfer, H. J. Chem. Phys. Lipids 1979, 24, 321–333. doi:10.1016/0009-3084(79)90117-8 |
| 16. | Scott, A. I.; Dodson, P. A.; McCapra, F.; Meyers, M. B. J. Am. Chem. Soc. 1963, 85, 3702–3704. doi:10.1021/ja00905a040 |
| 17. | Coutts, I. G. C.; Edwards, M.; Musto, D. R.; Richards, D. J. Tetrahedron Lett. 1980, 21, 5055–5056. doi:10.1016/S0040-4039(00)71131-2 |
| 18. | Coutts, I. G. C.; Culbert, N. J.; Edwards, M.; Hadfield, J. A.; Musto, D. R.; Pavlidis, V. H.; Richards, D. J. J. Chem. Soc., Perkin Trans. 1 1985, 1829–1836. doi:10.1039/P19850001829 |
| 19. | Thomas, H. G.; Schwager, H.-W. Tetrahedron Lett. 1984, 25, 4471–4474. doi:10.1016/S0040-4039(01)81469-6 |
| 20. | Deffieux, D.; Fabre, I.; Courseille, C.; Quideau, S. J. Org. Chem. 2002, 67, 4458–4465. doi:10.1021/jo020023r |
| 10. | Torii, S.; Tanaka, H. Carboxylic Acids. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; Marcel Dekker: New York, 2001; pp 499–543. |
| 11. | Schäfer, H. J. Recent Contributions of Kolbe Electrolysis to Organic Synthesis. In Electrochemistry IV; Steckhan, E., Ed.; Topics in Current Chemistry, Vol. 152; Springer-Verlag: New York, 1990; pp 91–151. |
| 21. | Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924 |
| 5. | Sutterer, A.; Moeller, K. D. J. Am. Chem. Soc. 2000, 122, 5636–5637. doi:10.1021/ja001063k |
| 9. | Campbell, J. M.; Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2012, 134, 18338–18344. doi:10.1021/ja307046j |
| 13. | Lebreux, F.; Buzzo, F.; Markó, I. E. Synlett 2008, 2815–2820. doi:10.1055/s-0028-1083547 |
| 14. | Huhtasaari, M.; Schäfer, H. J.; Becking, L. Angew. Chem., Int. Ed. Engl. 1984, 23, 980–981. doi:10.1002/anie.198409801 |
| 15. | Matzeit, A.; Schäfer, H. J.; Amatore, C. Synthesis 1995, 1432–1444. doi:10.1055/s-1995-4112 |
© 2013 Perkins et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)