Abstract
Water induces a change in the product of radical multifunctionalization reactions of aliphatic alkenes involving an sp3 C–H functionalization by an 1,5-hydrogen shift using tert-butyl nitrite and molecular oxygen. The reaction without water, reported previously, gives nitrated γ-lactols, whereas the reaction in the presence of water produces 4-hydroxy-5-nitropentyl nitrate or 4-hydroxy-3-nitropentyl nitrate derivatives.

Graphical Abstract
Introduction
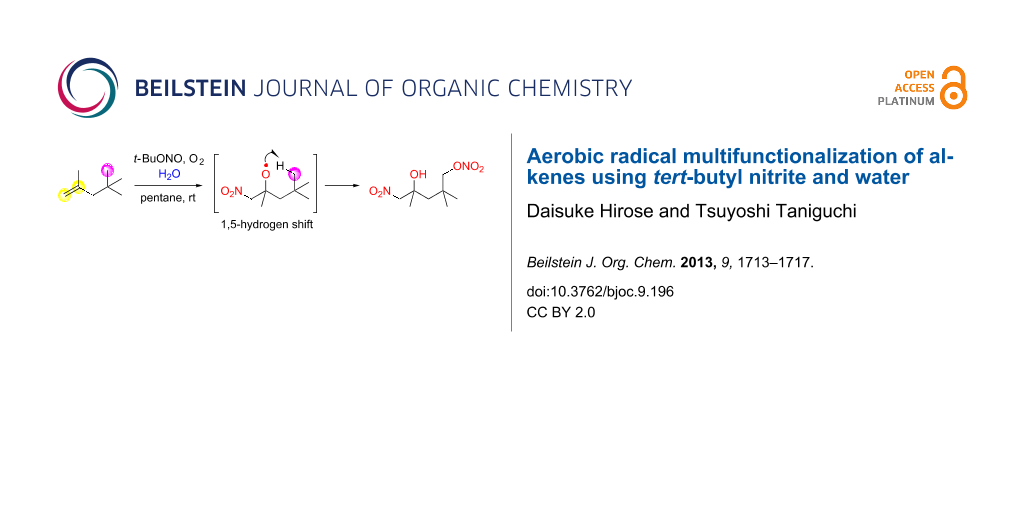
Multifunctionalization reactions of simple organic molecules are useful methods because they can provide a shortcut to desired products. Various methods used widely, such as organometallic reactions, cycloaddition reactions and multicomponent reactions, are utilized for multifunctionalization [1-5]. If direct functionalization reactions of inactivated bonds, such as simple multiple bonds or C–H bonds, are combined with multifunctionalization processes, they would become more efficient synthetic methods [6-9]. In this regard, radical reactions are capable of realizing multifunctionalization of inactivated organic molecules due to their high reactivity. Addition reactions of radical species to multiple bonds in the presence of appropriate trapping reagents give double functionalized compounds [10,11]. In addition, a radical methodology is a powerful tool for sp3 C–H functionalization [12-16]. It is frequently achieved by intramolecular radical hydrogen transfer reactions. An 1,5-hydrogen shift is the most favourable process [17], and useful methods such as the Barton reaction and the Hofmann–Löffler–Freytag reaction have been reported [18-20]. Recently, we reported a novel radical multifunctionalization reaction of aliphatic alkenes using tert-butyl nitrite (t-BuONO) and oxygen [21]. In this reaction, three positions including an unreactive sp3 C–H bond are functionalized in alkenes to produce nitrated γ-lactols in one step. For instance, treatment of alkene 1 with 5 equivalents of t-BuONO in a dried solvent under an oxygen atmosphere directly gave γ-lactol 2 (Scheme 1). On the other hand, we found in the course of this study that addition of water altered the products to a non-cyclic functionalized compound 3 (Scheme 1). In this letter, we report water-induced multifunctionalization reactions of alkenes by aerobic radical oxynitration.
![[1860-5397-9-196-i1]](/bjoc/content/inline/1860-5397-9-196-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The effect of water in radical multifunctionalization reactions.
Scheme 1: The effect of water in radical multifunctionalization reactions.
Results and Discussion
Our investigation started with the screening of solvents in the reaction of alkene 1 with t-BuONO (3 equiv) and oxygen in the presence of water (3 equiv). Although dimethyl sulfoxide (DMSO) was the best solvent in the reaction without water to obtain γ-lactol 2 [21], it worked inefficiently in the presence of water, and triple functionalized product 3 was obtained in low yield along with a large amount of oxynitration product 4 (Table 1, entry 1). The use of other polar solvents such as N,N-dimethylformamide (DMF) and tetrahydrofuran (THF) also did not give better results (Table 1, entries 2 and 3), whereas the use of dichloromethane (CH2Cl2) improved the ratio of 3 and 4 (ca. 1:2), though the isolated yield of 3 was still low (entry 4). Encouraged by this observation, we tested the reaction in pentane and obtained an improved ratio (ca. 1:1) and improved yield (26%) of 3 (entry 5). Eventually, the reaction with an excess of water (30 equiv) and with a longer reaction time (12 h) somewhat improved the yield (33%) of 3. This reaction was reproducible on a larger scale (6 mmol) and gave 3 in 37% yield. Water was insoluble in the organic solvent, but it seemed to cause no problem with the reproducibility. Although we tried to further optimize the conditions, such as amounts of reagents, concentration, temperature and sonication, no better results were obtained. Efforts for optimizations are still being continued.
Table 1: The effect of solvents.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-196-i4.svg?max-width=637&scale=1.0)
|
|||
| entry | solvent | yield (%)a | |
|---|---|---|---|
| 3 | 4 | ||
| 1 | DMSO | 15 | 54 |
| 2 | DMF | 15 | 43 |
| 3 | THF | 18 | 52 |
| 4 | CH2Cl2 | 13 | 28 |
| 5 | pentane | 26 | 28 |
| 6b | pentane | 33 (37)c | 37 (38)c |
aIsolated yield. b30 equiv of H2O, 12 h. c6.0 mmol scale.
Next, our interest focused on the multifunctionalization reactions of various alkenes 5–14 (Table 2). The oxynitration of olefins and the direct oxygenation of methyl C–H bonds of branched aliphatic alkenes 5–10 occurred to afford triple functionalized products 15–20 (Table 2, entries 1–6). In the reactions of alkenes 5 and 7, a remarkable diastereoselectivity was observed (Table 2, entries 1 and 3, and vide infra). We found that the multifunctionalization of monosubstituted alkene 8 took place to give 18 unlike the reaction without water [21], though the yield was not high (entry 4). The reaction of 1,1,3,3-tetramethyl-5-methylenecyclohexane (9) gave the mutifunctionalized product 19 as a single isomer. The stereochemistry of 19 could be presumably assigned by considering the 1,5-hydrogen shift mechanism (entry 5) [17]. The reaction of alkene 10 bearing a methyl ester moiety gave γ-lactone 20 by intramolecular transesterification with a tertiary hydroxy group arising from hydration of the olefin (entry 6). Functionalization reactions involving oxygenation of methylene C–H bonds of alkenes 11–13 also proceeded to afford secondary nitrate compounds 21–23, though no diastereoselectivity was observed (Table 2, entries 7–9). Unfortunately, no tertiary nitrate ester 24 was detected in the reaction of 14. This implies that the 1,5-hydrogen shift on the methine site is not so fast (entry 10). Typically, in the radical hydrogen abstraction, a methine C–H bond is more reactive than methyl and methylene sites, whereas it might be difficult for substrates having a methine group to form an organized transition state to induce the 1,5-hydrogen shift because they have only one hydrogen atom that can be abstracted [17]. In short, entropy factors might be dominant in the present reaction.
Table 2: Reactions of various alkenesa.
| entry | alkene | product | yieldb |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-196-i5.svg?max-width=637&scale=1.0)
5 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-196-i6.svg?max-width=637&scale=1.0)
15 |
40%; dr, 90:10 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-196-i7.svg?max-width=637&scale=1.0)
6 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-196-i8.svg?max-width=637&scale=1.0)
16 |
29% |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-196-i9.svg?max-width=637&scale=1.0)
7 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-196-i10.svg?max-width=637&scale=1.0)
17 |
38%; dr, >95:5 |
| 4c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-196-i11.svg?max-width=637&scale=1.0)
8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-196-i12.svg?max-width=637&scale=1.0)
18 |
16% |
| 5c |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-196-i13.svg?max-width=637&scale=1.0)
9 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-196-i14.svg?max-width=637&scale=1.0)
19 |
28% |
| 6b |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-196-i15.svg?max-width=637&scale=1.0)
10 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-196-i16.svg?max-width=637&scale=1.0)
20 |
31%; dr, 50:50 |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-196-i17.svg?max-width=637&scale=1.0)
11 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-196-i18.svg?max-width=637&scale=1.0)
21 |
31%; dr, 50:50 |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-196-i19.svg?max-width=637&scale=1.0)
12 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-196-i20.svg?max-width=637&scale=1.0)
22 |
33%; dr, 50:50 |
| 9 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-196-i21.svg?max-width=637&scale=1.0)
13 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-196-i22.svg?max-width=637&scale=1.0)
23 |
37%; dr, 50:50 |
| 10 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-196-i23.svg?max-width=637&scale=1.0)
14 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-196-i24.svg?max-width=637&scale=1.0)
24 |
not detected |
aReaction conditions: alkene (1.0 mmol), t-BuONO (3.0 mmol), H2O (30 mmol) in pentane (5 mL) under an oxygen atmosphere (balloon) for 12 h. bIsolated yield. Diastereomeric ratio (dr) was approximately estimated by 1H NMR analysis. c3 equiv (3.0 mmol) of H2O was used.
We attempted to transform the product obtained by the present reaction into a valuable compound. As a represent example, compound 3 was exposed to typical hydrogenation conditions using 10% palladium on carbon to obtain the corresponding 5-amino-1,4-diol compound. Since this is a highly polar compound, diol 25 was isolated after protection of the amino group by subsequent treatment of the crude product with di-tert-butyl dicarbonate (Boc2O) (Scheme 2). This experiment emphasised the potential of the present multifunctionalization method because the simple unsaturated hydrocarbon 1 can be converted into the highly polar amino-1,4-diol compound in only two steps.
![[1860-5397-9-196-i2]](/bjoc/content/inline/1860-5397-9-196-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Transformation of 3 into 5-amino-1,4-diol derivative 25.
Scheme 2: Transformation of 3 into 5-amino-1,4-diol derivative 25.
The plausible mechanism is fundamentally the same as the previously proposed one (Scheme 3) [21-23]. Certainly, the present reaction is triggered by addition of nitrogen dioxide (NO2) generated by aerobic decomposition of t-BuONO to the olefin of 1. After the resultant tertiary carbon radical has been trapped by molecular oxygen, an alkoxy radical 26 is formed from the peroxynitrite intermediate (ROONO) generated by the reaction of the peroxy radical (ROO•) intermediate with t-BuONO. The alkoxy radical causes an 1,5-hydrogen shift to give the corresponding alkyl radical 27 followed by formation of another alkoxy radical 28 through a similar process. An important issue in the current work is the effect of water to determine the destiny of 28. In the absence of water, the alkoxy radical 28 was merely oxidized to aldehyde 29, which was converted into γ-lactol 2 [21]. On the other hand, if 28 is caught by NO2 more rapidly than the oxidation, nitrate ester 3 is produced, and it was really observed in the presence of water. These observations clearly indicate that a difference in the concentration of NO2 in situ affects the results of each reaction. In other words, water may promote the generation of NO2 from t-BuONO, and this observation has already been reported in previous radical oxynitration reactions of alkenes [22]. In addition, when alkene 1 was subjected to nitrous acid (HNO2), that was formed from sodium nitrite (NaNO2; 3 equiv) and acetic acid (3 equiv), and oxygen in water (30 equiv) and pentene (0.2 M), production of compound 3 was observed, though the yield was low (7%). Since this system is an alternative method for the generation of NO2, this may support the formation of same intermediate species such as HNO2 and NO2 in the reaction using t-BuONO, oxygen and water [24].
![[1860-5397-9-196-i3]](/bjoc/content/inline/1860-5397-9-196-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The high diastereoselectivity in reactions of alkenes 5 and 7 could be rationalized by six-membered transition states 30 and 31 in the 1,5-hydrogen shift. If chair forms were postulated in both cases, more substituents should be arranged at equatorial positions to form more stable transition states. According to this presumption, the configurations of the major isomers of products 15 and 17 can be tentatively assigned as shown in Scheme 3.
Conclusion
We found in multifunctionalization reactions of alkenes with t-BuONO and oxygen that water induced the production of another triple functionalized compound that is a nitrate ester. This can be a precursor of a 5-amino-1,4-diol, and the access from simple aliphatic alkenes in one step is unprecedented. The functionalization of unreactive sp3 C–H bonds using a radical 1,5-hydrogen shift is an old methodology compared with modern transition metal-catalyzed C–H activation reactions [25-27], but the current work has shown that this old methodology still has a large potential. The development of other direct C–H functionalization reactions based on radical chemistry is currently on-going together with further optimization of the presented reaction in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data of all products, and copies of NMR spectra. | ||
| Format: PDF | Size: 1.8 MB | Download |
Acknowledgements
Authors thank Mr. Yuki Sugiura (Kanazawa University) for performing some preliminary experiments. Authors are also grateful to Drs. Shigeyoshi Kanoh, Katsuhiro Maeda, and Tomoyuki Ikai (Kanazawa University) for helpful discussions. This work was partially supported by a Grant-in-Aid for Scientific Research (C) (25460011) from The Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
-
Pellissier, H. Chem. Rev. 2013, 113, 442–524. doi:10.1021/cr300271k
Return to citation in text: [1] -
Matsuo, Y.; Isobe, H.; Tanaka, T.; Murata, Y.; Murata, M.; Komatsu, K.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 17148–17149. doi:10.1021/ja056077a
Return to citation in text: [1] -
Hilt, G.; Hess, W.; Harms, K. Org. Lett. 2006, 8, 3287–3290. doi:10.1021/ol061153f
Return to citation in text: [1] -
Jiang, B.; Tu, S.-J.; Kaur, P.; Wever, W.; Li, G. J. Am. Chem. Soc. 2009, 131, 11660–11661. doi:10.1021/ja904011s
Return to citation in text: [1] -
Arai, S.; Koike, Y.; Hada, H.; Nishida, A. J. Am. Chem. Soc. 2010, 132, 4522–4523. doi:10.1021/ja910451j
Return to citation in text: [1] -
Muñiz, K.; Martínez, C. J. Org. Chem. 2013, 78, 2168–2174. doi:10.1021/jo302472w
Return to citation in text: [1] -
Souto, J. A.; Becker, P.; Iglesias, Á.; Muñiz, K. J. Am. Chem. Soc. 2012, 134, 15505–15511. doi:10.1021/ja306211q
Return to citation in text: [1] -
Li, Y.-M.; Sun, M.; Wang, H.-L.; Tian, Q.-P.; Yang, S.-D. Angew. Chem., Int. Ed. 2013, 52, 3972–3976. doi:10.1002/anie.201209475
Return to citation in text: [1] -
Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M. Angew. Chem., Int. Ed. 2013, 52, 4000–4003. doi:10.1002/anie.201210250
Return to citation in text: [1] -
Renaud, P.; Sibi, M. P., Eds. Radicals in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2001. doi:10.1002/9783527618293
Return to citation in text: [1] -
Tojino, M.; Ryu, I. Free-Radical-Mediated Multicomponent Coupling Reactions. In Multicomponent Reactions; Zhu, J.; Bienaymé, H., Eds.; Wiley-VCH: Weinheim, Germany; pp 169–198. doi:10.1002/3527605118.ch6
Return to citation in text: [1] -
Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d
Return to citation in text: [1] -
Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k
Return to citation in text: [1] -
Ishihara, Y.; Baran, P. S. Synlett 2010, 1733–1745. doi:10.1055/s-0030-1258123
Return to citation in text: [1] -
Chen, H.; Sanjaya, S.; Wang, Y.-F.; Chiba, S. Org. Lett. 2013, 15, 212–215. doi:10.1021/ol303302r
Return to citation in text: [1] -
Amaoka, Y.; Nagatomo, M.; Inoue, M. Org. Lett. 2013, 15, 2160–2163. doi:10.1021/ol4006757
Return to citation in text: [1] -
Čeković, Ž. Tetrahedron 2003, 59, 8073–8090. doi:10.1016/S0040-4020(03)01202-X
Return to citation in text: [1] [2] [3] -
Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. J. Am. Chem. Soc. 1960, 82, 2640–2641. doi:10.1021/ja01495a061
Return to citation in text: [1] -
Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. J. Am. Chem. Soc. 1961, 83, 4076–4083. doi:10.1021/ja01480a030
Return to citation in text: [1] -
Wolff, M. E. Chem. Rev. 1963, 63, 55–64. doi:10.1021/cr60221a004
Return to citation in text: [1] -
Taniguchi, T.; Sugiura, Y.; Hatta, T.; Yajima, A.; Ishibashi, H. Chem. Commun. 2013, 49, 2198–2200. doi:10.1039/C3CC00130J
Return to citation in text: [1] [2] [3] [4] [5] -
Taniguchi, T.; Yajima, A.; Ishibashi, H. Adv. Synth. Catal. 2011, 353, 2643–2647. doi:10.1002/adsc.201100315
Return to citation in text: [1] [2] -
Maity, S.; Naveen, T.; Sharma, U.; Maiti, D. Org. Lett. 2013, 15, 3384–3387. doi:10.1021/ol401426p
Return to citation in text: [1] -
Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Tetrahedron 2010, 66, 9077–9106. doi:10.1016/j.tet.2010.09.057
Return to citation in text: [1] -
Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. doi:10.1002/anie.200806273
Return to citation in text: [1] -
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e
Return to citation in text: [1] -
Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j
Return to citation in text: [1]
| 1. | Pellissier, H. Chem. Rev. 2013, 113, 442–524. doi:10.1021/cr300271k |
| 2. | Matsuo, Y.; Isobe, H.; Tanaka, T.; Murata, Y.; Murata, M.; Komatsu, K.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 17148–17149. doi:10.1021/ja056077a |
| 3. | Hilt, G.; Hess, W.; Harms, K. Org. Lett. 2006, 8, 3287–3290. doi:10.1021/ol061153f |
| 4. | Jiang, B.; Tu, S.-J.; Kaur, P.; Wever, W.; Li, G. J. Am. Chem. Soc. 2009, 131, 11660–11661. doi:10.1021/ja904011s |
| 5. | Arai, S.; Koike, Y.; Hada, H.; Nishida, A. J. Am. Chem. Soc. 2010, 132, 4522–4523. doi:10.1021/ja910451j |
| 17. | Čeković, Ž. Tetrahedron 2003, 59, 8073–8090. doi:10.1016/S0040-4020(03)01202-X |
| 24. | Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Tetrahedron 2010, 66, 9077–9106. doi:10.1016/j.tet.2010.09.057 |
| 12. | Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d |
| 13. | Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800–3842. doi:10.1021/cr040170k |
| 14. | Ishihara, Y.; Baran, P. S. Synlett 2010, 1733–1745. doi:10.1055/s-0030-1258123 |
| 15. | Chen, H.; Sanjaya, S.; Wang, Y.-F.; Chiba, S. Org. Lett. 2013, 15, 212–215. doi:10.1021/ol303302r |
| 16. | Amaoka, Y.; Nagatomo, M.; Inoue, M. Org. Lett. 2013, 15, 2160–2163. doi:10.1021/ol4006757 |
| 25. | Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. doi:10.1002/anie.200806273 |
| 26. | Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e |
| 27. | Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j |
| 10. | Renaud, P.; Sibi, M. P., Eds. Radicals in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2001. doi:10.1002/9783527618293 |
| 11. | Tojino, M.; Ryu, I. Free-Radical-Mediated Multicomponent Coupling Reactions. In Multicomponent Reactions; Zhu, J.; Bienaymé, H., Eds.; Wiley-VCH: Weinheim, Germany; pp 169–198. doi:10.1002/3527605118.ch6 |
| 21. | Taniguchi, T.; Sugiura, Y.; Hatta, T.; Yajima, A.; Ishibashi, H. Chem. Commun. 2013, 49, 2198–2200. doi:10.1039/C3CC00130J |
| 6. | Muñiz, K.; Martínez, C. J. Org. Chem. 2013, 78, 2168–2174. doi:10.1021/jo302472w |
| 7. | Souto, J. A.; Becker, P.; Iglesias, Á.; Muñiz, K. J. Am. Chem. Soc. 2012, 134, 15505–15511. doi:10.1021/ja306211q |
| 8. | Li, Y.-M.; Sun, M.; Wang, H.-L.; Tian, Q.-P.; Yang, S.-D. Angew. Chem., Int. Ed. 2013, 52, 3972–3976. doi:10.1002/anie.201209475 |
| 9. | Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M. Angew. Chem., Int. Ed. 2013, 52, 4000–4003. doi:10.1002/anie.201210250 |
| 22. | Taniguchi, T.; Yajima, A.; Ishibashi, H. Adv. Synth. Catal. 2011, 353, 2643–2647. doi:10.1002/adsc.201100315 |
| 21. | Taniguchi, T.; Sugiura, Y.; Hatta, T.; Yajima, A.; Ishibashi, H. Chem. Commun. 2013, 49, 2198–2200. doi:10.1039/C3CC00130J |
| 17. | Čeković, Ž. Tetrahedron 2003, 59, 8073–8090. doi:10.1016/S0040-4020(03)01202-X |
| 21. | Taniguchi, T.; Sugiura, Y.; Hatta, T.; Yajima, A.; Ishibashi, H. Chem. Commun. 2013, 49, 2198–2200. doi:10.1039/C3CC00130J |
| 21. | Taniguchi, T.; Sugiura, Y.; Hatta, T.; Yajima, A.; Ishibashi, H. Chem. Commun. 2013, 49, 2198–2200. doi:10.1039/C3CC00130J |
| 22. | Taniguchi, T.; Yajima, A.; Ishibashi, H. Adv. Synth. Catal. 2011, 353, 2643–2647. doi:10.1002/adsc.201100315 |
| 23. | Maity, S.; Naveen, T.; Sharma, U.; Maiti, D. Org. Lett. 2013, 15, 3384–3387. doi:10.1021/ol401426p |
| 21. | Taniguchi, T.; Sugiura, Y.; Hatta, T.; Yajima, A.; Ishibashi, H. Chem. Commun. 2013, 49, 2198–2200. doi:10.1039/C3CC00130J |
| 18. | Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. J. Am. Chem. Soc. 1960, 82, 2640–2641. doi:10.1021/ja01495a061 |
| 19. | Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. J. Am. Chem. Soc. 1961, 83, 4076–4083. doi:10.1021/ja01480a030 |
| 20. | Wolff, M. E. Chem. Rev. 1963, 63, 55–64. doi:10.1021/cr60221a004 |
| 17. | Čeković, Ž. Tetrahedron 2003, 59, 8073–8090. doi:10.1016/S0040-4020(03)01202-X |
© 2013 Hirose and Taniguchi; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)