Abstract
Piperazirum, isolated from Arum palaestinum Boiss, was originally assigned as r-3,c-5-diisobutyl-c-6-isopropylpiperazin-2-one. The reported structure was synthesised diastereoselectively using a key nitro-Mannich reaction to set up the C5/C6 relative stereochemistry. The structure was unambiguously assigned by single crystal X-ray diffraction but the spectroscopic data did not match those reported for the natural product. The structure of the natural product must therefore be revised.

Graphical Abstract
Introduction
The nitro-Mannich reaction (or aza-Henry reaction) has been developed to a standard where the product β-nitroamines 1 are now privileged building blocks. In part this is due to the complementary synthetic flexibility available from the two different nitrogen atom oxidation states (Scheme 1) [1]. They have been used to synthesise many nitrogen-containing functional groups including α-amino carbonyls [2,3], peptidomimetics [4], natural products [5-10] and many heterocyclic small molecules [11-24] of importance to drug discovery. Enantioselective reactions have been controlled by asymmetric metal-centred Lewis acids; chiral hydrogen bond donors, in particular by the use of asymmetric thiourea organocatalysts, chiral Brønsted acids, phase-transfer catalysts and Brønsted base catalysts [3,15,25-39]. A large part of our own work in developing the nitro-Mannich reaction was to demonstrate the preparation of stereodefined 1,2-diamines [40-45].
![[1860-5397-9-200-i1]](/bjoc/content/inline/1860-5397-9-200-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Schematic nitro-Mannich reaction.
Scheme 1: Schematic nitro-Mannich reaction.
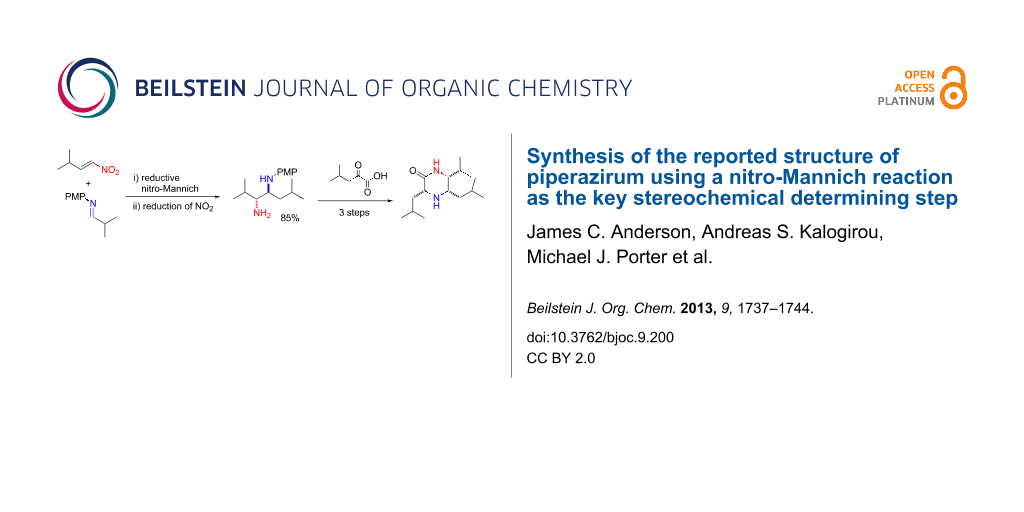
As part of a programme aimed at using these 1,2-diamines as building blocks in target synthesis we focused on the synthesis of a novel bioactive alkaloid, piperazirum (2, Scheme 2), which to our knowledge had not been previously synthesised. Piperazirum was isolated from the leaf extract of Arum palaestinum Boiss and was shown to possess significant cytotoxicity against cultured tumor cell lines in vitro [46]. Its chemical structure and relative stereochemistry were elucidated by high resolution mass spectrometry, infrared, 1D and 2D NMR spectroscopy [46]. Retrosynthetically we envisaged that the C-3 stereocentre could be set up by hydrogenation from the less hindered face of α-iminolactam 3 or α,β-unsaturated lactam 4 (Scheme 2). These heterocycles could be derived from a common α-keto acid derivative 5 and either diamine 6 or 7, that could in turn be prepared from β-nitroamine 8 or 9. Each of the β-nitroamines could be prepared enantioselectively by using our previously reported methodology [28,43,45] and would allow elucidation of the absolute stereochemistry of piperazirum (2).
![[1860-5397-9-200-i2]](/bjoc/content/inline/1860-5397-9-200-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic analysis of piperazirum.
Scheme 2: Retrosynthetic analysis of piperazirum.
Results and Discussion
The common α-keto acid derivative 5 was easily prepared from a Grignard reaction of isobutylmagnesium chloride with diethyl oxalate to give α-keto ester 10 in 94% yield (Scheme 3) [47]. Saponification of 10 with KOH provided α-keto acid 11 in excellent yield [48], and the corresponding acid chloride 12 was prepared in situ by treatment with oxalyl chloride [49].
![[1860-5397-9-200-i3]](/bjoc/content/inline/1860-5397-9-200-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: (a) iBuMgCl, Et2O, −78 °C; (b) KOH, EtOH/H2O, 100 °C; (c) (COCl)2.
Scheme 3: (a) iBuMgCl, Et2O, −78 °C; (b) KOH, EtOH/H2O, 100 °C; (c) (COCl)2.
For the synthesis of β-nitroamine 8 we decided to make use of the reductive nitro-Mannich reaction as the starting nitroalkene 13 is readily available from a Henry reaction [50], imine 14 from the condensation of p-anisidine and isobutyraldehyde, and the process can easily be made asymmetric [45]. Conjugate addition of hydride to 13 and subsequent trapping of the nitronate anion with freshly prepared imine 14 in THF gave β-nitroamine 15 in 64% conversion and a dr of 70:30 (Scheme 4). Quite frequently β-nitroamines are unstable and susceptible to retroaddition [43,44]. Formation of the corresponding trifluoroacetamide derivative confers stability and allows them to be purified. Using previously developed conditions to protect the amine in situ using (CF3CO)2O gave only β-nitroamine 16 as a single diastereoisomer in a low 15% yield. These results are consistent with the poor conversions and dr, as well as resistance to trifluoroacetamide protection, we have observed before from imines derived from α-branched aldehydes such as cyclohexanecarbaldehyde [44].
![[1860-5397-9-200-i4]](/bjoc/content/inline/1860-5397-9-200-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (a) Li(Et3BH), THF, rt then 14, CF3CO2H, −78 °C, dr 70:30; (b) (CF3CO)2O, Py, CH2Cl2, 0 °C to rt; (c) Zn, 6 M HCl, EtOAc/EtOH, rt.
Scheme 4: (a) Li(Et3BH), THF, rt then 14, CF3CO2H, −78 °C, dr 70:30; (b) (CF3CO)2O, Py, CH2Cl2, 0 °C to rt; (...
In cases where trifluoroacetylation fails, it is quite common to purify the β-nitroamine by rapid column chromatography, quickly followed by reduction to give the corresponding 1,2-diamine. In this case rapid purification of 15 followed by reduction with Zn/HCl gave the 1,2-diamine 17 as a single diastereoisomer in 50% yield (Scheme 4).
With diamine 17 in hand, the reaction with a suitable keto acid derivative was investigated. We presumed that the nitrogen of the primary amine would be the more nucleophilic and hence should react with the more electrophilic carbonyl group – i.e. the ketone – in 10 [51,52]. Attempted formation of 4 by heating a mixture of 10 and 17 in H2O at 50 °C gave a complicated mixture of products and attempts under Dean–Stark conditions in toluene with TsOH gave only recovered starting materials [53].
In light of this poor result, the alternative route to 2 via β-nitroamine 9 was investigated. A reductive nitro-Mannich reaction between nitroalkene 18 [54] and freshly prepared imine 19 in CH2Cl2 followed by rapid flash chromatography gave β-nitroamine 20 with complete conversion and dr >95:5 [55]. As before immediate reduction with Zn/HCl gave the PMP-protected diamine 21 in 85% yield as a single diastereoisomer determined by 1H NMR (Scheme 5).
![[1860-5397-9-200-i5]](/bjoc/content/inline/1860-5397-9-200-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: (a) Li(Et3BH), CH2Cl2, rt then 19, CF3CO2H, −78 °C, dr >95:5; (b) Zn, 6 M HCl, EtOAc/EtOH, rt, single diastereoisomer; (c) CSCl2, aq NaHCO3 CH2Cl2/ MeOH, rt.
Scheme 5: (a) Li(Et3BH), CH2Cl2, rt then 19, CF3CO2H, −78 °C, dr >95:5; (b) Zn, 6 M HCl, EtOAc/EtOH, rt, sing...
From a comprehensive series of examples of the reductive nitro-Mannich reaction, the vast majority of substrates demonstrate anti-relative stereochemistry [23,44,45]. More direct proof for 21 was gleaned from the corresponding imidazolidine-2-thione formed by treatment with thiophosgene to give 22 (Scheme 5). In one dimensional nOe studies irradiation of the CHNH peak (δ 3.70, 1H, dd, J = 8.4, 5.4 Hz) caused a 3.65% enhancement of the CHN peak (δ 4.31, 1H, dt, J = 7.9, 5.9 Hz), indicating a cis-relative stereochemistry between the two protons, which confirmed the anti-relative stereochemistry of 21. The observed coupling constant between the same two protons was 8.2 Hz (averaged) and was similar to other imidazolidine-2-thiones we have prepared that have been corroborated by single crystal X-ray crystallography [56]. Further stereochemical proof was provided by a single crystal X-ray structure determination of 2·HCl (vide infra).
We presumed again that the primary amine of 21 would be more nucleophilic towards a keto acid derivative 5. In order to obtain a piperazinone of the desired connectivity (23) a keto acid derivative would be required where the carboxylate carbonyl is more reactive than the ketone carbonyl. Two possible such compounds were considered, acid chloride 12 and carboxylic acid 11 treated with a suitable coupling agent. Acid chloride 12 was prepared in situ by treatment of acid 11 with oxalyl chloride (2.00 equiv) and catalytic DMF. Subsequent reaction with diamine 21 in the presence of pyridine (1.20 equiv) and catalytic DMAP over 24 h, according to previously reported reactions for similar keto acids [49], gave only the bis-adduct 24 and none of the desired piperazinone 23. By contrast, the reaction of carboxylic acid 11 with diamine 21, in the presence of EDC (1.50 equiv) and 1-hydroxybenzotriazole (1.50 equiv) at rt, gave the desired product 23 in good yield (Scheme 6) [57]. The double bond geometry was assigned as Z by NOESY 1H NMR and probably results from steric inhibition of resonance which would result in the formation of the E iPr group and the planar amide group during formation of 23.
![[1860-5397-9-200-i6]](/bjoc/content/inline/1860-5397-9-200-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: (a) (COCl)2, DMF, CH2Cl2, rt; (b) 21, Py, DMAP, CH2Cl2, rt; (c) EDC, HOBt, THF/CH2Cl2, rt; (d) H2 (1 atm), Pd/C, MeOH, rt; (e) CAN, MeCN/H2O, 0 °C; (f) 4 M HCl in dioxane, MeCN/Et2O.
Scheme 6: (a) (COCl)2, DMF, CH2Cl2, rt; (b) 21, Py, DMAP, CH2Cl2, rt; (c) EDC, HOBt, THF/CH2Cl2, rt; (d) H2 (...
Reduction of the double bond with hydrogen over palladium on charcoal gave a single diastereoisomer 25 in quantitative yield. In one dimensional nOe studies irradiation of H-3 (δ 4.09, 1H, dd, J = 9.9, 4.5 Hz) caused a 0.17% enhancement of the H-6 signal (δ 3.18, 1H, dd, J = 10.0, 3.5 Hz) with negligible enhancement of the H-5 signal (δ 3.37, 1H, dt, J = 12.4, 3.1 Hz). Irradiation of H-5 caused a 2.90% enhancement of the H-6 signal and a 0.07% enhancement of the H-3 signal. The nOe data tentatively suggested that protons H-3, H-5 and H-6 were all on the same face of the molecule. Deprotection of the PMP group with CAN gave compound 2. Extensive NMR (COSY, HMBC and NOESY) and analytical data were consistent with the structure drawn (2), but the 1H and 13C NMR did not match that published for piperazirum (Table 1) [46]. The original authors recorded their NMR data for piperazirum in D2O [58], but our sample 2 was insoluble. Compound 2 was readily soluble in DMSO-d6 and CDCl3, but neither gave a satisfactory match to the reported NMR data. Preparation of 2·HCl allowed the recording of NMR spectra in D2O, but again the chemical shifts were inconsistent with those reported. Single crystal X-ray structure determination of 2 proved unambiguously the assigned structure obtained from spectroscopic data (Figure 1) [59].
Table 1: Comparison of selected 13C NMR chemical shifts of piperazirum, 2 and 2·HCl.
|
Carbon
atom |
piperazirum δc (ppm)
D2O [49] |
2 δc (ppm)
DMSO-d6 |
2 δc (ppm)
CDCl3 |
2·HCl δc (ppm)
D2O |
|---|---|---|---|---|
| C-2 | 175.7 | 172.5 | 174.3 | 169.9 |
| C-3 | 53.6 | 56.4 | 56.9 | 54.9 |
| C-5 | 59.7 | 53.2 | 53.3 | 53.9 |
| C-6 | 60.6 | 58.1 | 59.4 | 56.3 |
| CH2C-3 | 40.0 | 41.5 | 41.1 | 38.8 |
| CH2C-5 | 24.6 | 40.2 | 40.5 | 36.3 |
![[1860-5397-9-200-1]](/bjoc/content/figures/1860-5397-9-200-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of 2·HCl.
Figure 1: X-ray crystal structure of 2·HCl.
Conclusion
The rapid synthesis of r-3,c-5-diisobutyl-c-6-isopropylpiperazin-2-one has been achieved using an efficient nitro-Mannich reaction to establish the C-5/C-6 relative stereochemistry which in turn controls the formation of the stereocentre at C-3. Spectrosocpic and single crystal X-ray data have shown that the reported structure for piperazirum is erroneous and that the structure of the natural product needs to be revised. While the reported data point strongly towards piperazirum having the same connectivity as 2, it is not clear which of the three alternative diastereomers corresponds to the natural product. In view of the lack of natural material, further chemical synthesis, guided by GIAO chemical shift prediction, is currently underway in an effort to elucidate the correct structure for piperazirum. In addition the determination of further biological activity of 2 and its diastereoisomers will be investigated.
Experimental
Unless otherwise stated, all reactions were carried out under an atmosphere of nitrogen. All glassware was flame dried and allowed to cool under a stream of nitrogen before use. Cooling to 0 °C was effected using an ice–water bath. Cooling to temperatures below 0 °C was effected using dry ice–acetone mixtures. Reactions were monitored by thin layer chromatography (TLC) using 0.25 mm silica precoated plastic plates with fluorescent indicator. Sheets were visualised using ultraviolet light (254 nm) and/or anisaldehyde or KMnO4 solutions, as appropriate. Removal of solvents (in vacuo) was achieved using a water aspirator and rotary evaporators. Flash column chromatography was performed using silica gel 60, 40–63 μm. Commercial solvents and reagents were used as supplied or purified in accordance with standard procedures [60]. Diethyl ether (Et2O), tetrahydrofuran (THF), dichloromethane (CH2Cl2) and toluene (PhMe) were obtained from a solvent tower, where degassed solvent was passed through two columns of activated alumina and a 7 micron filter under 4 bar pressure. All NMR samples were made as dilute solutions of CDCl3 unless otherwise stated. All chemical shifts (δ) are reported in parts per million (ppm) relative to residual solvent peaks except in D2O where they are relative to dioxane (D2O) 1H 3.75 ppm and 13C 67.2 ppm. Multiplicities for 1H coupled signals are denoted as s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet. Coupling constants (J) are reported in Hertz. 13C multiplicities were assigned using a DEPT sequence. Where appropriate, HMQC, COSY, HMBC and NOE experiments were carried out to aid assignment. Melting points are uncorrected.
(E)-4-Methoxy-N-(3-methylbutylidene)aniline (19): To a mixture of p-anisidine (123 mg, 1.00 mmol) and basic alumina (1.00 g) in CH2Cl2 (5 mL) at −78 °C was added isovaleraldehyde (107 µL, 1.00 mmol) and the mixture stirred for 1 h, then warmed to rt, filtered and evaporated in vacuo to give crude imine 19 (182 mg, 95%) as a colourless oil that was used immediately without further purification; 1H NMR (600 MHz) δ 1.02 (d, J = 6.7, 6H, CH3), 2.04 (sept, J = 6.7, 1H, CH(CH3)2), 2.34 (dd, J = 7.0, 5.4, 2H, CH2), 3.80 (s, 1H, OCH3), 6.87 (app. d, J = 8.9, 2H, ArH), 7.02 (app. d, J = 8.9, 2H, ArH), 7.86 (t, J = 5.4, 1H, N=CH); 13C NMR (150 MHz) data were in agreement to that reported [61].
(3R*,4S*)-N4-(4-Methoxyphenyl)-2,6-dimethylheptane-3,4-diamine (21): To a solution of nitroalkene 18 (202 mg, 2.00 mmol), in CH2Cl2 (12.0 mL) was added Superhydride® (2.20 mL, 1 M in THF, 2.20 mmol) and the mixture stirred for 15 min at rt. The mixture was cooled to −78 °C before the dropwise addition of a solution of freshly prepared imine 19 (564 mg, 4.00 mmol) in CH2Cl2 (12.0 mL). The reaction was stirred for 10 min before the dropwise addition of a solution of CF3CO2H (460 µL, 6.00 mmol) in CH2Cl2 (4.0 mL). The reaction was stirred for 1 h and then quenched with brine (20 mL) at −78 °C, warmed to rt and extracted with Et2O (3 × 20 mL). The combined organics were dried (MgSO4) and evaporated in vacuo to give crude β-nitroamine 20, that was purified by column chromatography (petrol ether/Et2O 4:1). Subsequent reaction with 6 M HCl (6.60 mL, 40.0 mmol) and Zn dust (1.30 g, 20.0 mmol) and purification by flash column chromatography (CH2Cl2/MeOH 10:1) gave diamine 21 (452 mg, 85%) as a brown oil; Rf 0.50 (CH2Cl2/MeOH 10:1); IR νmax (thin film): 3374 (br, N-H), 2955 (w, C-H), 1618 (w), 1508 (s, C=C), 1465 (m), 1441 (w), 1385 (w), 1366 (w), 1292 (w), 1238 (s), 1179 (w), 1154 (w), 1038 (m), 816 (m), 752 (s) cm−1; 1H NMR (600 MHz) δ 0.90 (d, J = 6.5, 3H, CH3), 0.92 (d, J = 6.7, 3H, CH3), 0.99 (d, J = 6.6, 6H, CH3), 1.21 (m, 1H, CH2), 1.35 (m, 1H, CH2), 1.59 (m, 1H, CH(CH3)2), 1.80 (m, 1H, CH(CH3)2), 2.53 (dd, J = 9.1, 2.7, 1H, CHNH2), 3.52 (m, 1H, CHNH), 3.73 (s, 3H, OCH3), 6.57 (app. d, J = 8.9, 2H, ArH), 6.76 (app. d, J = 8.9, 2H, ArH); 13C NMR (150 MHz) δ 19.6 (CH3), 20.6 (CH3), 21.7 (CH3), 24.1 (CH3), 24.7 (CH(CH3)2), 31.4 (CH(CH3)2), 37.2 (CH2), 52.9 (CHNH), 55.8 (OCH3), 59.1 (CHNH2), 114.4 (CH arom.), 115.0 (CH arom.), 142.3 (Cq arom.), 151.6 (Cq arom.); MS (EI+) m/z: 264 (M+, 5%), 192 (M+ − (CH3)2CHCHNH2, 100%); HRMS m/z: calcd for C16H28N2O, 264.2196; found, 264.2201.
(5S*,6R*,Z)-5-Isobutyl-6-isopropyl-4-(4-methoxyphenyl)-3-(2-methylpropylidene)piperazin-2-one (23): To a solution of diamine 21 (61 mg, 0.23 mmol) in THF (5 mL) was added N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 67 mg, 0.35 mmol) and 1-hydroxybenzotriazole hydrate (47 mg, 0.35 mmol) followed by a solution of keto acid 11 (30 mg, 0.23 mmol) in CH2Cl2 (5 mL) at rt. The mixture was stirred for 24 h, then diluted with CH2Cl2 (20 mL) and washed with brine (10 mL). The combined organic phases were dried (MgSO4), evaporated in vacuo and purified by flash column chromatography (petrol ether/EtOAc 7:3) to give piperazinone 23 (57 mg, 69%) as a brown solid; mp 152–153 °C; Rf 0.30 (petrol ether/EtOAc 7:3); IR νmax (thin film): 3209 (br, N-H), 2956 (w, C-H), 1671 (s, C=C), 1622 (s), 1499 (s), 1464 (m), 1442 (m), 1409 (m), 1384 (m), 1366 (m), 1331 (m), 1283 (m), 1241 (s), 1180 (m), 1153 (m), 1037 (m), 826 (s), 767 (m) cm−1; 1H NMR (600 MHz) δ 0.68 (d, J = 6.6, 3H, CH3), 0.79 (d, J = 6.6, 3H, CH3), 0.88 (d, J = 6.6, 3H, CH3), 0.93 (d, J = 6.5, 3H, CH3), 0.95 (d, J = 6.7, 3H, CH3), 1.07 (d, J = 6.5, 3H, CH3), 1.07 (m, 1H, CH2), 1.55 (m, 1H, CH2), 1.55 (m, 1H, CH(CH3)2), 1.94 (m, 1H, CH(CH3)2), 2.43 (m, 1H, CH(CH3)2), 3.18 (dd, J = 10.3, 3.6, 1H, CHNH), 3.53 (br. d, J = 12.2, 1H, NCHCH2), 3.76 (s, 3H, OCH3), 5.89 (br. s, 1H, NH), 6.48 (d, J = 10.6, 1H, =CH), 6.78 (app. d, J = 8.8, 2H, ArH), 6.89 (app. d, J = 8.8, 2H, ArH); 13C NMR (150 MHz) δ 17.9 (CH3), 19.4 (CH3), 20.3 (CH3), 21.5 (CH3), 22.2 (CH3), 23.7 (CH3), 24.1 (CH(CH3)2), 26.4 (CH(CH3)2), 29.0 (CH(CH3)2), 34.4 (CH2), 55.4 (OCH3), 58.0 (CHNH), 58.8 (NCHCH2), 114.3 (CH arom.), 122.0 (CH arom.), 129.7 (CH=Cq), 140.0 (Cq=CH), 143.4 (Cq arom.), 154.3 (Cq arom.), 164.3 (C=O); MS (EI+) m/z: 359 (MH+, 100%), 343 (12%), 192 (13%); HRMS m/z: calcd for C22H35N2O2, 359.2693; found, 359.2678.
(3R*,5S*,6R*)-3,5-diisobutyl-6-isopropyl-4-(4-methoxyphenyl)piperazin-2-one (25): To a solution of piperazinone 23 (170 mg, 0.470 mmol) in MeOH (10 mL) was added palladium on carbon (50 mg, 10% by weight, 0.047 mmol) and the mixture was flushed with hydrogen, then stirred under a hydrogen atmosphere (balloon) at rt. After the piperazinone starting material was consumed (TLC, 4 h) the mixture was filtered through celite®, washed with CH2Cl2 (20 mL) and evaporated in vacuo to give crude piperazinone that was purified by flash column chromatography (petrol ether/Me2CO 4:1) to give piperazinone 25 (170 mg, 99%) as a colourless oil; Rf 0.50 (petrol ether/Me2CO 4:1); IR νmax (thin film): 3207 (br, N-H), 2954 (m, C-H), 1658 (s, C=O), 1505 (C=C), 1465 (m), 1367 (m), 1242 (s), 1180 (m), 1039 (m), 827 (m), 788 (m), 733 (m) cm−1; 1H NMR (600 MHz) δ 0.70 (d, J = 6.5, 3H, CH3), 0.77 (d, J = 6.7, 3H, CH3), 0.90 (d, J = 6.7, 3H, CH3), 0.97 (d, J = 6.5, 3H, CH3), 0.98 (d, J = 6.8, 3H, CH3), 1.03 (d, J = 6.5, 3H, CH3), 1.03 (m, 1H, CHCHCH2), 1.55 (m, 1H, CHCHCH2), 1.55 (m, 1H, O=CCHCH2), 1.55 (m, 1H, CHCH(CH3)2), 1.84 (m, 1H, O=CCHCH2), 1.92 (m, 1H, O=CCHCH2CH(CH3)2), 2.06 (m, 1H, CHCHCH2CH(CH3)2), 3.18 (dd, J = 10.0, 3.5, 1H, NHCH), 3.37 (dt, J = 12.4, 3.1, 1H, NHCHCHN), 3.77 (s, 3H, OCH3), 4.09 (dd, J = 9.9, 4.5, 1H, O=CCH), 6.02 (br. s, 1H, NH), 6.80 (app. d, J = 8.9, 2H, ArH), 6.91 (app. d, J = 8.9, 2H, ArH); 13C NMR (150 MHz) δ 18.0 (CH3), 19.5 (CH3), 21.5 (CH3), 21.6 (CH3), 23.3 (CH3), 23.4 (CHCHCH2CH(CH3)2), 23.7 (CH3), 24.7 (O=CCHCH2CH(CH3)2), 29.2 (NHCHCH(CH3)2), 34.7 (CH2CHCH), 44.0 (CH2CHC=O), 55.4 (OCH3), 57.2 (NCHC=O), 58.4 (NCHCH), 58.5 (NHCH), 114.5 (CH arom.), 122.6 (CH arom.), 146.2 (CH arom.), 154.3 (CH arom.), 173.9 (C=O); MS (EI+) m/z: 360 (M+, 15%), 303 (18%), 192 (100%); HRMS m/z: calcd for C22H36N2O2, 360.2771; found, 360.2774.
(3R*,5S*,6R*)-3,5-diisobutyl-6-isopropylpiperazin-2-one (2): To a solution of piperazinone 25 (320 mg, 0.880 mmol) in MeCN (10 mL) at 0 °C was added a solution of CAN (2.08 g, 3.52 mmol) in H2O (10 mL) dropwise over 3 min. The solution turned from pale yellow to dark orange. The mixture was stirred at 0 °C for 2 h, over which time the solution became light orange. Water (30 mL) was then added and the mixture extracted with EtOAc (3 × 20 mL), washed with saturated aqueous NaHCO3 (40 mL), dried (MgSO4) and evaporated in vacuo to give crude piperazinone that was purified by flash column chromatography (petrol ether/Me2CO 3:2) to give piperazinone 2 (91 mg, 41%) as a brown oil; Rf 0.53 (petrol ether/Me2CO 3:2); IR νmax (thin film): 3209 (w, N-H), 2955 (C-H), 1658 (s, C=O), 1467 (m), 1367 (m), 1165 (w), 918 (w), 722 (w) cm−1; 1H NMR (600 MHz, CDCl3) δ 0.89 (d, J = 6.6, 3H, C-3CH2CHCH3), 0.90 (d, J = 6.2, 3H, C-5CH2CHCH3), 0.91 (d, J = 5.6, 3H, C-6CHCH3), 0.93 (d, J = 6.9, 3H, C-3CH2CHCH3), 0.94 (d, J = 6.8, 3H, C-5CH2CHCH3), 0.98 (d, J = 6.7, 3H, C-6CHCH3), 1.30 (ddd, J = 13.9, 7.1, 6.5, 1H, C-5CH2), 1.33 (ddd, J = 13.9, 8.2, 5.6, 1H, C-5CH2), 1.40 (ddd, J = 14.2, 10.0, 4.1, 1H, C-3CH2), 1.65 (nonet, J = 6.7, 1H, C-5CH2CH), 1.74 (m, 1H, C-3CH2CH), 1.88 (ddd, J = 13.7, 10.3, 3.3, 1H, C-3CH2), 1.91 (hepd, J = 6.8, 2.5, 1H, C-6CH), 3.06 (dd, J = 6.7, 3.6, 1H, C-6H), 3.15 (dt, J = 7.8, 5.3, 1H, C-5H), 3.40 (dd, J = 10.2, 3.4, 1H, C-3H), 6.22 (brs, 1H, N1H), N4H peak is missing; 13C NMR (125 MHz, CDCl3) δ 17.7 (C-6CHCH3), 21.0 (C-3CH2CHCH3), 21.5 (C-5CH2CHCH3), 22.4 (C-6CHCH3), 23.1 and 23.7 (C-3CH2CHCH3/ C-5CH2CHCH3), 24.4 (C-3CH2CH), 24.8 (C-5CH2CH), 27.9 (C-6CH), 40.5 (C-5CH2), 41.1 (C-3CH2), 53.3 (C-5), 57.0 (C-3), 59.4 (C-6), 174.2 (C=O); MS (EI+) m/z: 254 (M+, 30%), 197 (22%), 169 (43%), 154 (31%); HRMS m/z: calcd for C15H30N2O, 254.2353; found, 254.2355.
1H NMR (600 MHz, DMSO-d6) δ 0.83–0.89 (m, 18H, 6xMe), 1.25 (m, 3H, C-3CH2 + 2xC-5CH2), 1.65 (m, 2H, C-3CH2CH + C-5CH2CH), 1.79 (m, 2H, C-3CH2 + C-6CH), 2.90 (brd, J = 3.2, 1H, C-6H), 3.01 (td, J = 7.1, 3.7, 1H, C-5H), 3.21 (dd, J = 9.3, 3.2, 1H, C-3H), 7.63 (brs, 1H, NH). Assignments based on above; 13C NMR (125 MHz, DMSO-d6) δ 18.4, 21.5, 22.0, 22.8, 23.0, 23.8, 23.9, 24.2, 27.4, 40.2, 41.5, 53.2, 56.4, 58.1, 172.5.
In a clean vial HCl in dioxane (~1 equiv) was added to a small sample of 2 in MeCN, the mixture layered with Et2O and then left in the fridge until colourless blades of 2·HCl had formed at the interface. mp 121–123 °C. 1H NMR (600 MHz, D2O) δ 0.89–0.99 (6xMe), 1.52–1.68 (m, 4H), 1.72–1.79 (m, 1H), 1.94–1.99 (m, 2H), 3.50 (dd, J = 6.2, 4.3, 1H, C-6H), 3.79 (m, 1H, C-5H), 4.01 (dd, J = 8.3, 5.3, 1H, C-3H); 13C NMR (150 MHz, D2O) δ 18.8, 20.6, 21.3, 22.0, 22.5, 24.2, 24.7, 25.7, 27.4, 36.3, 38.8, 53.9, 54.9, 56.3, 169.9 (C=O).
Supporting Information
| Supporting Information File 1: Further experimental and characterisation data. | ||
| Format: PDF | Size: 111.8 KB | Download |
References
-
Noble, A.; Anderson, J. C. Chem. Rev. 2013, 113, 2887. doi:10.1021/cr300272t
See for a comprehensive review of the nitro-Mannich reaction.
Return to citation in text: [1] -
Ballini, R.; Petrini, M. Tetrahedron 2004, 60, 1017. doi:10.1016/j.tet.2003.11.016
Return to citation in text: [1] -
Palomo, C.; Oiarbide, M.; Halder, R.; Laso, A.; López, R. Angew. Chem., Int. Ed. 2006, 45, 117. doi:10.1002/anie.200502674
Return to citation in text: [1] [2] -
Bursavich, M. G.; Rich, D. H. J. Med. Chem. 2002, 45, 541. doi:10.1021/jm010425b
Return to citation in text: [1] -
Shen, B.; Johnston, J. N. Org. Lett. 2008, 10, 4397. doi:10.1021/ol801797h
Return to citation in text: [1] -
Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632. doi:10.1021/ja908399s
Return to citation in text: [1] -
Jakubec, P.; Kyle, A. F.; Calleja, J.; Dixon, D. J. Tetrahedron Lett. 2011, 52, 6094. doi:10.1016/j.tetlet.2011.09.016
Return to citation in text: [1] -
Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037. doi:10.1039/c1cc13665h
Return to citation in text: [1] -
Kumaraswamy, G.; Pitchaiah, A. Helv. Chim. Acta 2011, 94, 1543. doi:10.1002/hlca.201100013
Return to citation in text: [1] -
Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D. J. J. Am. Chem. Soc. 2012, 134, 17482. doi:10.1021/ja308826x
Return to citation in text: [1] -
Tsuritani, N.; Yamada, K.-I.; Yoshikawa, N.; Shibasaki, M. Chem. Lett. 2002, 31, 276. doi:10.1246/cl.2002.276
Return to citation in text: [1] -
Xu, X.; Furukawa, T.; Okino, T.; Miyabe, H.; Takemoto, Y. Chem.–Eur. J. 2006, 12, 466. doi:10.1002/chem.200500735
Return to citation in text: [1] -
Anderson, J. C.; Chapman, H. A. Org. Biomol. Chem. 2007, 5, 2413–2422. doi:10.1039/b705081j
Return to citation in text: [1] -
Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389. doi:10.1021/ol800108u
Return to citation in text: [1] -
Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. J. J. Am. Chem. Soc. 2010, 132, 4925. doi:10.1021/ja100514y
Return to citation in text: [1] [2] -
Weng, J.; Li, Y.-B.; Wang, R.-B.; Li, F.-Q.; Liu, C.; Chan, A. S. C.; Lu, G. J. Org. Chem. 2010, 75, 3125. doi:10.1021/jo100187m
Return to citation in text: [1] -
Kumaraswamy, G.; Pitchaiah, A. Tetrahedron 2011, 67, 2536. doi:10.1016/j.tet.2011.02.031
Return to citation in text: [1] -
Davis, T. A.; Johnston, J. N. Chem. Sci. 2011, 2, 1076. doi:10.1039/c1sc00061f
Return to citation in text: [1] -
Xie, H.; Zhang, Y.; Zhang, S.; Chen, X.; Wang, W. Angew. Chem., Int. Ed. 2011, 50, 11773. doi:10.1002/anie.201105970
Return to citation in text: [1] -
Jensen, K. L.; Poulsen, P. H.; Donslund, B. S.; Morana, F.; Jørgensen, K. A. Org. Lett. 2012, 14, 1516. doi:10.1021/ol3002514
Return to citation in text: [1] -
Anderson, J. C.; Horsfall, L. R.; Kalogirou, A. S.; Mills, M. R.; Stepney, G. J.; Tizzard, G. J. J. Org. Chem. 2012, 77, 6186–6198. doi:10.1021/jo301000r
Return to citation in text: [1] -
Davis, T. A.; Danneman, M. W.; Johnston, J. N. Chem. Commun. 2012, 48, 5578. doi:10.1039/c2cc32225k
Return to citation in text: [1] -
Anderson, J. C.; Noble, A.; Tocher, D. A. J. Org. Chem. 2012, 77, 6703. doi:10.1021/jo3010827
Return to citation in text: [1] [2] -
Anderson, J. C.; Noble, A.; Ribelles Torres, P. Tetrahedron Lett. 2012, 53, 5707. doi:10.1016/j.tetlet.2012.08.062
Return to citation in text: [1] -
Marquéz-López, E.; Merino, P.; Tejero, T.; Herrera, R. P. Eur. J. Org. Chem. 2009, 2401. doi:10.1002/ejoc.200801097
See for a review specific to asymmetric methods.
Return to citation in text: [1] -
Yamada, K.-I.; Moll, G.; Shibasaki, M. Synlett 2001, 980. doi:10.1055/s-2001-14639
Return to citation in text: [1] -
Knudsen, K. R.; Risgaard, T.; Nishiwaki, N.; Gothelf, K. V.; Jørgensen, K. A. J. Am. Chem. Soc. 2001, 123, 5843. doi:10.1021/ja010588p
Return to citation in text: [1] -
Anderson, J. C.; Howell, G. P.; Lawrence, R. M.; Wilson, C. S. J. Org. Chem. 2005, 70, 5665. doi:10.1021/jo050762i
Return to citation in text: [1] [2] -
Trost, B. M.; Lupton, D. W. Org. Lett. 2007, 9, 2023. doi:10.1021/ol070618e
Return to citation in text: [1] -
Chen, Z.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2008, 130, 2170. doi:10.1021/ja710398q
Return to citation in text: [1] -
Yamada, K.-I.; Harwood, S. J.; Gröger, H.; Shibasaki, M. Angew. Chem., Int. Ed. 1999, 38, 3504. doi:10.1002/(SICI)1521-3773(19991203)38:23<3504::AID-ANIE3504>3.0.CO;2-E
Return to citation in text: [1] -
Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 4900. doi:10.1021/ja0701560
Return to citation in text: [1] -
Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Org. Lett. 2004, 6, 625. doi:10.1021/ol0364531
Return to citation in text: [1] -
Nugent, B. M.; Yoder, R. A.; Johnston, J. N. J. Am. Chem. Soc. 2004, 126, 3418. doi:10.1021/ja031906i
Return to citation in text: [1] -
Yoon, T. P.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2005, 44, 466. doi:10.1002/anie.200461814
Return to citation in text: [1] -
Fini, F.; Sgarzani, V.; Petterson, D.; Herrera, R. P.; Bernardi, L.; Ricci, A. Angew. Chem., Int. Ed. 2005, 44, 7975. doi:10.1002/anie.200502646
Return to citation in text: [1] -
Palomo, C.; Oiarbide, M.; Laso, A.; López, R. J. Am. Chem. Soc. 2005, 127, 17622. doi:10.1021/ja056594t
Return to citation in text: [1] -
Robak, M. T.; Trincado, M.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 15110. doi:10.1021/ja075653v
Return to citation in text: [1] -
Wang, C.-J.; Dong, X.-Q.; Zhang, Z.-H.; Xue, Z.-Y.; Teng, H.-L. J. Am. Chem. Soc. 2008, 130, 8606. doi:10.1021/ja803538x
Return to citation in text: [1] -
Adams, H.; Anderson, J. C.; Peace, S.; Pennell, A. M. K. J. Org. Chem. 1998, 63, 9932. doi:10.1021/jo981700d
Return to citation in text: [1] -
Anderson, J. C.; Peace, S.; Pih, S. Synlett 2000, 850. doi:10.1055/s-2000-6710
Return to citation in text: [1] -
Anderson, J. C.; Blake, A. J.; Howell, G. P.; Wilson, C. J. Org. Chem. 2005, 70, 549. doi:10.1021/jo048304h
Return to citation in text: [1] -
Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961. doi:10.1021/jo102408u
Return to citation in text: [1] [2] [3] -
Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711. doi:10.1021/jo300535h
Return to citation in text: [1] [2] [3] [4] -
Anderson, J. C.; Koovits, P. J. Chem. Sci. 2013, 4, 2897. doi:10.1039/c3sc50613d
Return to citation in text: [1] [2] [3] [4] -
El-Desouky, S.-K.; Ryu, S. Y.; Kim, Y.-K. Tetrahedron Lett. 2007, 48, 4015. doi:10.1016/j.tetlet.2007.04.032
Return to citation in text: [1] [2] [3] -
Rambaud, M.; Bakasse, M.; Duguay, G.; Villieras, J. Synthesis 1988, 564. doi:10.1055/s-1988-27642
Return to citation in text: [1] -
Ramage, R.; Griffiths, G. J.; Shutt, F. E.; Sweeney, J. N. A. J. Chem. Soc., Perkin Trans. 1 1984, 1531. doi:10.1039/P19840001531
Return to citation in text: [1] -
Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j
Return to citation in text: [1] [2] [3] -
Kumaran, G.; Kulkarni, G. H. Synthesis 1995, 1545. doi:10.1055/s-1995-4137
Return to citation in text: [1] -
Boeykens, M.; De Kimpe, N.; Tehrani, K. A. J. Org. Chem. 1994, 59, 6973. doi:10.1021/jo00102a022
Return to citation in text: [1] -
Li, L.-S.; Zhou, Y.; Zhao, J.; Dragovich, P. S.; Stankovic, N.; Bertolini, T. M.; Murphy, D. E.; Sun, Z.; Tran, C. V.; Ayida, B. K.; Ruebsam, F.; Webber, S. E. Synthesis 2007, 3301. doi:10.1055/s-2007-990823
Return to citation in text: [1] -
Murthy, S. N.; Madhav, B.; Nageswar, Y. V. D. Helv. Chim. Acta 2010, 93, 1216. doi:10.1002/hlca.200900358
Return to citation in text: [1] -
Lambert, A.; Lowe, A. J. Chem. Soc. 1947, 1517. doi:10.1039/jr9470001517
Return to citation in text: [1] -
Reaction in THF gave a lower stereoselectivity of dr 85:15. The use of CH2Cl2 as solvent in reductive nitro-Mannich reactions can give enhanced diastereoselectivities. See [24].
Return to citation in text: [1] -
Kaliogirou, A. S. Ph.D. Thesis, University College London, London, 2013.
Return to citation in text: [1] -
Grijalvo, S.; Bedia, C.; Triola, G.; Casas, J.; Llebaria, A.; Teixidó, J.; Rabal, O.; Levade, T.; Delgado, A.; Fabriàs, G. Chem. Phys. Lipids 2006, 144, 69. doi:10.1016/j.chemphyslip.2006.07.001
Return to citation in text: [1] -
The publication [49] states in the discussion that the NMR were recorded in DMSO-d6, but is then tabulated in D2O. The author confirmed the data was recorded in D2O and that there were no suitable samples of the material left for re-analysis and the original spectra were unobtainable. Private communication from Prof Young-Kyoon Kim.
Return to citation in text: [1] -
Crystallographic data (excluding structure factors) for the structure of 2·HCl has been deposited with the Cambridge Crystallographic Data Centre under supplementary publication numbers CCDC 949530. Copies of the data can be obtained, free of charge, upon request from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or email: deposit@ccdc.cam.ac.uk].
Return to citation in text: [1] -
Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R. Purification of Laboratory Chemicals, 2nd ed.; Pergamon Press Ltd., 1980.
Return to citation in text: [1] -
Oiima, I.; Habus, H.; Zhao, M.; Zucco, M.; Park, Y. H.; Sun, C. M.; Brigaud, T. Tetrahedron 1992, 48, 6985. doi:10.1016/S0040-4020(01)91210-4
Return to citation in text: [1]
| 59. | Crystallographic data (excluding structure factors) for the structure of 2·HCl has been deposited with the Cambridge Crystallographic Data Centre under supplementary publication numbers CCDC 949530. Copies of the data can be obtained, free of charge, upon request from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or email: deposit@ccdc.cam.ac.uk]. |
| 49. | Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j |
| 60. | Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R. Purification of Laboratory Chemicals, 2nd ed.; Pergamon Press Ltd., 1980. |
| 1. |
Noble, A.; Anderson, J. C. Chem. Rev. 2013, 113, 2887. doi:10.1021/cr300272t
See for a comprehensive review of the nitro-Mannich reaction. |
| 11. | Tsuritani, N.; Yamada, K.-I.; Yoshikawa, N.; Shibasaki, M. Chem. Lett. 2002, 31, 276. doi:10.1246/cl.2002.276 |
| 12. | Xu, X.; Furukawa, T.; Okino, T.; Miyabe, H.; Takemoto, Y. Chem.–Eur. J. 2006, 12, 466. doi:10.1002/chem.200500735 |
| 13. | Anderson, J. C.; Chapman, H. A. Org. Biomol. Chem. 2007, 5, 2413–2422. doi:10.1039/b705081j |
| 14. | Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389. doi:10.1021/ol800108u |
| 15. | Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. J. J. Am. Chem. Soc. 2010, 132, 4925. doi:10.1021/ja100514y |
| 16. | Weng, J.; Li, Y.-B.; Wang, R.-B.; Li, F.-Q.; Liu, C.; Chan, A. S. C.; Lu, G. J. Org. Chem. 2010, 75, 3125. doi:10.1021/jo100187m |
| 17. | Kumaraswamy, G.; Pitchaiah, A. Tetrahedron 2011, 67, 2536. doi:10.1016/j.tet.2011.02.031 |
| 18. | Davis, T. A.; Johnston, J. N. Chem. Sci. 2011, 2, 1076. doi:10.1039/c1sc00061f |
| 19. | Xie, H.; Zhang, Y.; Zhang, S.; Chen, X.; Wang, W. Angew. Chem., Int. Ed. 2011, 50, 11773. doi:10.1002/anie.201105970 |
| 20. | Jensen, K. L.; Poulsen, P. H.; Donslund, B. S.; Morana, F.; Jørgensen, K. A. Org. Lett. 2012, 14, 1516. doi:10.1021/ol3002514 |
| 21. | Anderson, J. C.; Horsfall, L. R.; Kalogirou, A. S.; Mills, M. R.; Stepney, G. J.; Tizzard, G. J. J. Org. Chem. 2012, 77, 6186–6198. doi:10.1021/jo301000r |
| 22. | Davis, T. A.; Danneman, M. W.; Johnston, J. N. Chem. Commun. 2012, 48, 5578. doi:10.1039/c2cc32225k |
| 23. | Anderson, J. C.; Noble, A.; Tocher, D. A. J. Org. Chem. 2012, 77, 6703. doi:10.1021/jo3010827 |
| 24. | Anderson, J. C.; Noble, A.; Ribelles Torres, P. Tetrahedron Lett. 2012, 53, 5707. doi:10.1016/j.tetlet.2012.08.062 |
| 45. | Anderson, J. C.; Koovits, P. J. Chem. Sci. 2013, 4, 2897. doi:10.1039/c3sc50613d |
| 5. | Shen, B.; Johnston, J. N. Org. Lett. 2008, 10, 4397. doi:10.1021/ol801797h |
| 6. | Jakubec, P.; Cockfield, D. M.; Dixon, D. J. J. Am. Chem. Soc. 2009, 131, 16632. doi:10.1021/ja908399s |
| 7. | Jakubec, P.; Kyle, A. F.; Calleja, J.; Dixon, D. J. Tetrahedron Lett. 2011, 52, 6094. doi:10.1016/j.tetlet.2011.09.016 |
| 8. | Kyle, A. F.; Jakubec, P.; Cockfield, D. M.; Cleator, E.; Skidmore, J.; Dixon, D. J. Chem. Commun. 2011, 47, 10037. doi:10.1039/c1cc13665h |
| 9. | Kumaraswamy, G.; Pitchaiah, A. Helv. Chim. Acta 2011, 94, 1543. doi:10.1002/hlca.201100013 |
| 10. | Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D. J. J. Am. Chem. Soc. 2012, 134, 17482. doi:10.1021/ja308826x |
| 43. | Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961. doi:10.1021/jo102408u |
| 44. | Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711. doi:10.1021/jo300535h |
| 4. | Bursavich, M. G.; Rich, D. H. J. Med. Chem. 2002, 45, 541. doi:10.1021/jm010425b |
| 49. | Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j |
| 2. | Ballini, R.; Petrini, M. Tetrahedron 2004, 60, 1017. doi:10.1016/j.tet.2003.11.016 |
| 3. | Palomo, C.; Oiarbide, M.; Halder, R.; Laso, A.; López, R. Angew. Chem., Int. Ed. 2006, 45, 117. doi:10.1002/anie.200502674 |
| 46. | El-Desouky, S.-K.; Ryu, S. Y.; Kim, Y.-K. Tetrahedron Lett. 2007, 48, 4015. doi:10.1016/j.tetlet.2007.04.032 |
| 47. | Rambaud, M.; Bakasse, M.; Duguay, G.; Villieras, J. Synthesis 1988, 564. doi:10.1055/s-1988-27642 |
| 49. | Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j |
| 46. | El-Desouky, S.-K.; Ryu, S. Y.; Kim, Y.-K. Tetrahedron Lett. 2007, 48, 4015. doi:10.1016/j.tetlet.2007.04.032 |
| 48. | Ramage, R.; Griffiths, G. J.; Shutt, F. E.; Sweeney, J. N. A. J. Chem. Soc., Perkin Trans. 1 1984, 1531. doi:10.1039/P19840001531 |
| 40. | Adams, H.; Anderson, J. C.; Peace, S.; Pennell, A. M. K. J. Org. Chem. 1998, 63, 9932. doi:10.1021/jo981700d |
| 41. | Anderson, J. C.; Peace, S.; Pih, S. Synlett 2000, 850. doi:10.1055/s-2000-6710 |
| 42. | Anderson, J. C.; Blake, A. J.; Howell, G. P.; Wilson, C. J. Org. Chem. 2005, 70, 549. doi:10.1021/jo048304h |
| 43. | Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961. doi:10.1021/jo102408u |
| 44. | Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711. doi:10.1021/jo300535h |
| 45. | Anderson, J. C.; Koovits, P. J. Chem. Sci. 2013, 4, 2897. doi:10.1039/c3sc50613d |
| 61. | Oiima, I.; Habus, H.; Zhao, M.; Zucco, M.; Park, Y. H.; Sun, C. M.; Brigaud, T. Tetrahedron 1992, 48, 6985. doi:10.1016/S0040-4020(01)91210-4 |
| 3. | Palomo, C.; Oiarbide, M.; Halder, R.; Laso, A.; López, R. Angew. Chem., Int. Ed. 2006, 45, 117. doi:10.1002/anie.200502674 |
| 15. | Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. J. J. Am. Chem. Soc. 2010, 132, 4925. doi:10.1021/ja100514y |
| 25. |
Marquéz-López, E.; Merino, P.; Tejero, T.; Herrera, R. P. Eur. J. Org. Chem. 2009, 2401. doi:10.1002/ejoc.200801097
See for a review specific to asymmetric methods. |
| 26. | Yamada, K.-I.; Moll, G.; Shibasaki, M. Synlett 2001, 980. doi:10.1055/s-2001-14639 |
| 27. | Knudsen, K. R.; Risgaard, T.; Nishiwaki, N.; Gothelf, K. V.; Jørgensen, K. A. J. Am. Chem. Soc. 2001, 123, 5843. doi:10.1021/ja010588p |
| 28. | Anderson, J. C.; Howell, G. P.; Lawrence, R. M.; Wilson, C. S. J. Org. Chem. 2005, 70, 5665. doi:10.1021/jo050762i |
| 29. | Trost, B. M.; Lupton, D. W. Org. Lett. 2007, 9, 2023. doi:10.1021/ol070618e |
| 30. | Chen, Z.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2008, 130, 2170. doi:10.1021/ja710398q |
| 31. | Yamada, K.-I.; Harwood, S. J.; Gröger, H.; Shibasaki, M. Angew. Chem., Int. Ed. 1999, 38, 3504. doi:10.1002/(SICI)1521-3773(19991203)38:23<3504::AID-ANIE3504>3.0.CO;2-E |
| 32. | Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 4900. doi:10.1021/ja0701560 |
| 33. | Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Org. Lett. 2004, 6, 625. doi:10.1021/ol0364531 |
| 34. | Nugent, B. M.; Yoder, R. A.; Johnston, J. N. J. Am. Chem. Soc. 2004, 126, 3418. doi:10.1021/ja031906i |
| 35. | Yoon, T. P.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2005, 44, 466. doi:10.1002/anie.200461814 |
| 36. | Fini, F.; Sgarzani, V.; Petterson, D.; Herrera, R. P.; Bernardi, L.; Ricci, A. Angew. Chem., Int. Ed. 2005, 44, 7975. doi:10.1002/anie.200502646 |
| 37. | Palomo, C.; Oiarbide, M.; Laso, A.; López, R. J. Am. Chem. Soc. 2005, 127, 17622. doi:10.1021/ja056594t |
| 38. | Robak, M. T.; Trincado, M.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 15110. doi:10.1021/ja075653v |
| 39. | Wang, C.-J.; Dong, X.-Q.; Zhang, Z.-H.; Xue, Z.-Y.; Teng, H.-L. J. Am. Chem. Soc. 2008, 130, 8606. doi:10.1021/ja803538x |
| 28. | Anderson, J. C.; Howell, G. P.; Lawrence, R. M.; Wilson, C. S. J. Org. Chem. 2005, 70, 5665. doi:10.1021/jo050762i |
| 43. | Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961. doi:10.1021/jo102408u |
| 45. | Anderson, J. C.; Koovits, P. J. Chem. Sci. 2013, 4, 2897. doi:10.1039/c3sc50613d |
| 24. | Anderson, J. C.; Noble, A.; Ribelles Torres, P. Tetrahedron Lett. 2012, 53, 5707. doi:10.1016/j.tetlet.2012.08.062 |
| 53. | Murthy, S. N.; Madhav, B.; Nageswar, Y. V. D. Helv. Chim. Acta 2010, 93, 1216. doi:10.1002/hlca.200900358 |
| 44. | Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711. doi:10.1021/jo300535h |
| 51. | Boeykens, M.; De Kimpe, N.; Tehrani, K. A. J. Org. Chem. 1994, 59, 6973. doi:10.1021/jo00102a022 |
| 52. | Li, L.-S.; Zhou, Y.; Zhao, J.; Dragovich, P. S.; Stankovic, N.; Bertolini, T. M.; Murphy, D. E.; Sun, Z.; Tran, C. V.; Ayida, B. K.; Ruebsam, F.; Webber, S. E. Synthesis 2007, 3301. doi:10.1055/s-2007-990823 |
| 46. | El-Desouky, S.-K.; Ryu, S. Y.; Kim, Y.-K. Tetrahedron Lett. 2007, 48, 4015. doi:10.1016/j.tetlet.2007.04.032 |
| 58. | The publication [49] states in the discussion that the NMR were recorded in DMSO-d6, but is then tabulated in D2O. The author confirmed the data was recorded in D2O and that there were no suitable samples of the material left for re-analysis and the original spectra were unobtainable. Private communication from Prof Young-Kyoon Kim. |
| 49. | Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j |
| 57. | Grijalvo, S.; Bedia, C.; Triola, G.; Casas, J.; Llebaria, A.; Teixidó, J.; Rabal, O.; Levade, T.; Delgado, A.; Fabriàs, G. Chem. Phys. Lipids 2006, 144, 69. doi:10.1016/j.chemphyslip.2006.07.001 |
| 23. | Anderson, J. C.; Noble, A.; Tocher, D. A. J. Org. Chem. 2012, 77, 6703. doi:10.1021/jo3010827 |
| 44. | Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711. doi:10.1021/jo300535h |
| 45. | Anderson, J. C.; Koovits, P. J. Chem. Sci. 2013, 4, 2897. doi:10.1039/c3sc50613d |
| 55. | Reaction in THF gave a lower stereoselectivity of dr 85:15. The use of CH2Cl2 as solvent in reductive nitro-Mannich reactions can give enhanced diastereoselectivities. See [24]. |
© 2013 Anderson et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)