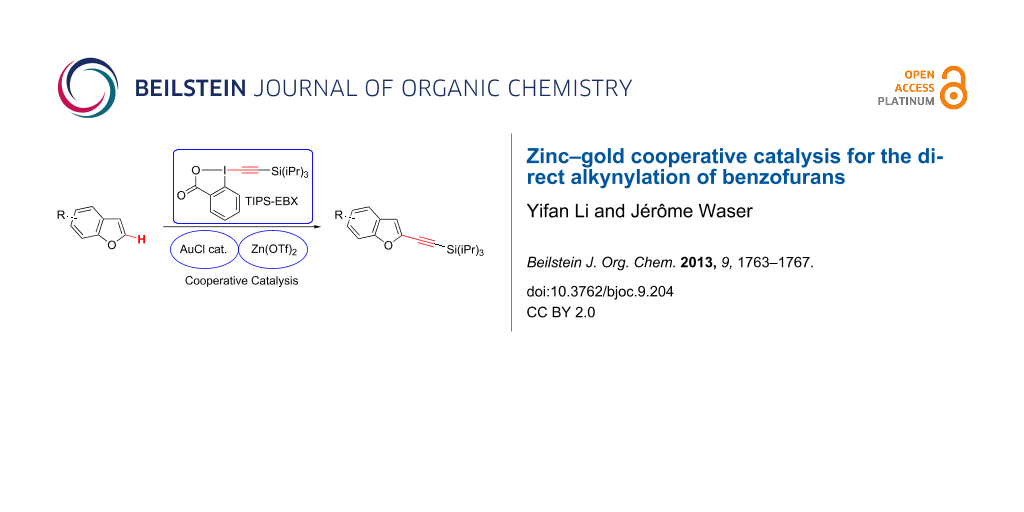
Abstract
The direct alkynylation of benzofurans was achieved for the first time using the hypervalent iodine reagent 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX) based on the cooperative effect between a gold catalyst and a zinc Lewis acid. High selectivity was observed for C2-alkynylation of benzofurans substituted with alkyl, aryl, halogen and ether groups. The reaction was also successful in the case of the more complex drug 8-methoxypsoralen (8-MOP).

Graphical Abstract
Introduction
Benzofurans are important heterocycles frequently encountered in both bioactive compounds and organic materials (Figure 1). For example, members of the furocoumarin class of natural products including psoralen (1), 8-methoxypsoralen (2) and angelicin (3) can cross-link with DNA upon light irradiation. They have consequently been used for the treatment of skin diseases such as cancer or psoriasis [1-4]. The natural product coumestrol (4) is found especially in soy beans and has estrogenic activity [5]. Synthetic bioactive compounds containing benzofurans are also important, as exemplified by amiodarone (5), as antiarrythmic drug [6,7]. Finally, benzofurans have also emerged recently as important structural elements for organic materials, such as the organic transistor 6 [8].
![[1860-5397-9-204-1]](/bjoc/content/figures/1860-5397-9-204-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Benzofurans in bioactive compounds and materials.
Figure 1: Benzofurans in bioactive compounds and materials.
Due to the importance of benzofurans, the discovery of new efficient methods for their synthesis and functionalization is an intensive field of research [9-11]. Especially interesting would be methods allowing the direct and regioselective C–H functionalization of benzofurans [12]. In this context, the introduction of an alkyne would be particularly useful, as acetylenes are important building blocks in synthetic chemistry, chemical biology and materials science [13]. Nevertheless, to the best of our knowledge, the direct alkynylation of benzofurans is still an unknown process.
Since 2009, our group has developed a mild gold-catalyzed [14-17] method for the alkynylation of electron-rich aryls such as indoles and pyrroles [18], thiophenes [19], anilines [20] and furans [21]. Key for success was the use of ethynylbenziodoxolones, which are cyclic hypervalent iodine reagents [22,23]. Nevertheless, the conditions we have used for other heterocycles gave only very low yields in the case of benzofurans. Herein, we would like to report the first catalytic direct C2-alkynylation of benzofurans 7 based on a cooperative effect between a gold catalyst and a zinc Lewis acid using 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX, 8) as reagent (Scheme 1). The reaction proceeded under mild conditions (60 °C under air) and could also be used to alkynylate the more complex polycyclic natural product 8-methoxypsoralen (2).
![[1860-5397-9-204-i1]](/bjoc/content/inline/1860-5397-9-204-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Zinc–gold catalyzed C2-alkynylation of benzofurans.
Scheme 1: Zinc–gold catalyzed C2-alkynylation of benzofurans.
Findings
Benzofuran (7a) is less reactive then furans and indeed no product was observed under the conditions optimized for the latter [21] at room temperature or at 60 °C using the commercially available electrophilic alkynylation reagent TIPS-EBX (8) (Table 1, entries 1 and 2) [24-27]. Fortunately, benzofuran (7a) was also more stable in the presence of acidic additives, and co-activation became possible, whereas Zn(OTf)2 was superior to trifluoroacetic acid (TFA) at 60 °C (Table 1, entries 3 and 4) [19,28]. No product was observed in the absence of AuCl, demonstrating the cooperative effect of the two metals (Table 1, entry 5). Lower or higher temperatures did not increase the yield (Table 1, entries 6 and 7). Finally, using a larger excess of TIPS-EBX (8) and Zn(OTf)2 gave 75% yield of alkynylation product 9a (Table 1, entry 8). The use of Zn(OTf)2 in catalytic amount led to a lower yield (Table 1, entry 9), and a larger excess resulted in decomposition of the starting material only (Table 1, entry 10). Although other Lewis acids could also be used (Table 1, entries 11 and 12) [29], no better results than with Zn(OTf)2 were obtained (Table 1, entry 7). Importantly, in contrast to our previous work with benzothiophenes [19], high selectivity for C2 alkynylation was observed.
Table 1: Optimization of the alkynylation of benzofuran (7a).
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-204-i5.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Equiv 8 | Additiveb | T [°C] | Yield |
|---|---|---|---|---|
| 1 | 1.2 | – | 23 | <5% |
| 2 | 1.2 | – | 60 | <5% |
| 3 | 1.2 | TFA | 60 | 42% |
| 4 | 1.2 | Zn(OTf)2 | 60 | 56% |
| 5 | 1.2 | Zn(OTf)2c | 60 | <5% |
| 6 | 1.2 | Zn(OTf)2 | 40 | 48% |
| 7 | 1.2 | Zn(OTf)2 | 82 | 36% |
| 8 | 2 | Zn(OTf)2 | 60 | 75% |
| 9 | 2 | Zn(OTf)2d | 60 | 37% |
| 10 | 2 | Zn(OTf)2e | 60 | 0% |
| 11 | 2 | Zn(NTf)2 | 60 | 57% |
| 12 | 2 | Yb(OTf)3 | 60 | 62% |
aReaction conditions: 7a (0.20 mmol) and AuCl (0.01 mmol) in acetonitrile (0.8 mL) under air for 26 h, isolated yield; bsame amount as 8; cwithout gold catalyst; d0.2 equiv; e4.0 equiv.
The scope of the reaction was then investigated (Scheme 2). Substitution by diverse functional groups was first examined on the C5 position. An electron-rich methoxy group was well tolerated, giving the desired alkynylation product 9b in 73% yield. The reaction was also successful with a bromide substituent (product 9c), making the method orthogonal to classical cross-coupling chemistry [30]. In presence of an aryl or an alkyl substituent, alkynylation was also obtained in 72% and 50% respectively (products 9d and 9e). Benzofurans substituted at the C7 position could also be used, as demonstrated by the efficient formation of alkynes 9f and 9g. In contrast, when 7-methoxybenzofuran (7j) was used, no C2 alkynylation product could be isolated. Instead, a mixture of C4 and C6 alkynylated benzofurans 9j and 9j’ was obtained (Scheme 3) [31]. Substitution on the furan ring was also possible at the C3 position (product 9h), but the use of 2-methylbenzofuran (7i) led to very a low yield in the alkynylation reaction.
![[1860-5397-9-204-i2]](/bjoc/content/inline/1860-5397-9-204-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-204-i3]](/bjoc/content/inline/1860-5397-9-204-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Alkynylation of 7-methoxybenzofuran (7j) [31].
Scheme 3: Alkynylation of 7-methoxybenzofuran (7j) [31].
Finally, we wondered if the alkynylation method could also be successful in the case of more complex benzofuran-containing natural products and drugs. We were pleased to see that the alkynylation of 8-methoxypsoralen (2) was indeed possible. The major product 10 bearing the acetylene group at the C5’ position was obtained in 37% yield (Scheme 4) [32]. Although the yield was still moderate, this was one of the first examples of direct alkynylation of a marketed drug. It also gave access in a single step to an interesting furocoumarin derivative with an extended chromophore, which could be important for phototherapy.
![[1860-5397-9-204-i4]](/bjoc/content/inline/1860-5397-9-204-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Alkynylation of 8-methoxypsoralen (2).
Scheme 4: Alkynylation of 8-methoxypsoralen (2).
Mechanistically, the reaction could proceed either via π-activation of the triple bond by the gold catalyst followed by conjugate addition of the benzofuran, α-elimination and 1,2-shift, or oxidative addition of TIPS-EBX (8) onto the gold catalyst (either at the Au(I) or Au(0) oxidation level) followed by electrophilic auration and reductive elimination [33]. The role of the zinc Lewis acid is not completely clear at this stage, but it may act by complexing the carboxylate group of the hypervalent iodine reagent, enhancing its electrophilic reactivity [19,34]. In fact, a complete shift of the 1H NMR signals of TIPS-EBX (8) was observed when Zn(OTf)2 was added, whereas no signal shift was observed when mixing the Lewis acid and benzofuran (7a) [35].
In conclusion, the first direct alkynylation method of benzofurans has been developed. Key for success was a cooperative effect between a gold catalyst and a zinc Lewis acid, together with the use of the hypervalent iodine reagent TIPS-EBX (8). Preliminary results obtained with 8-methoxypsoralen (2) demonstrated that the reaction could also be applied to more complex furocoumarin natural products.
Experimental
General procedure for the alkynylation of benzofurans: TIPS-EBX (8, 342 mg, 0.800 mmol, 2.0 equiv), AuCl (4.6 mg, 0.020 mmol, 0.050 equiv), Zn(OTf)2 (289 mg, 0.800 mmol, 2.0 equiv) and benzofuran 7 (0.40 mmol, 1.0 equiv) were added into CH3CN (2.0 mL) under air. The mixture was stirred for 26 hours at 60 °C. Then the mixture was concentrated in presence of silica gel and purified directly by column chromatography.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Song, P.-S.; Tapley, K. J., Jr. Photochem. Photobiol. 1979, 29, 1177. doi:10.1111/j.1751-1097.1979.tb07838.x
Return to citation in text: [1] -
Gasparro, F. P.; Dallamico, R.; Goldminz, D.; Simmons, E.; Weingold, D. Yale J. Biol. Med. 1989, 62, 579.
Return to citation in text: [1] -
Caffieri, S. Photochem. Photobiol. Sci. 2002, 1, 149. doi:10.1039/b107329j
Return to citation in text: [1] -
Santana, L.; Uriarte, E.; Roleira, F.; Milhazes, N.; Borges, F. Curr. Med. Chem. 2004, 11, 3239. doi:10.2174/0929867043363721
Return to citation in text: [1] -
Bickoff, E. M.; Booth, A. N.; Lyman, R. L.; Livingston, A. L.; Thompson, C. R.; Deeds, F. Science 1957, 126, 969. doi:10.1126/science.126.3280.969-a
Return to citation in text: [1] -
Mason, J. W. N. Engl. J. Med. 1987, 316, 455. doi:10.1056/NEJM198702193160807
Return to citation in text: [1] -
Bogazzi, F.; Tomisti, L.; Bartalena, L.; Aghini-Lombardi, F.; Martino, E. J. Endocrinol. Invest. 2012, 35, 340. doi:10.3275/8298
Return to citation in text: [1] -
Mitsui, C.; Soeda, J.; Miwa, K.; Tsuji, H.; Takeya, J.; Nakamura, E. J. Am. Chem. Soc. 2012, 134, 5448. doi:10.1021/ja2120635
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G.; Goggiomani, A. Heterocycles 2002, 56, 613. doi:10.3987/REV-01-SR(K)1
Return to citation in text: [1] -
Hou, X.-L.; Yang, Z.; Yeung, K.-S.; Wong, H. N. C. Five-membered ring systems: furans and benzofurans; Progress in Heterocyclic Chemistry, Vol. 18; Elsevier, 2007; pp 187–217. doi:10.1016/S0959-6380(07)80011-0
Return to citation in text: [1] -
De Luca, L.; Nieddu, G.; Porcheddu, A.; Giacomelli, G. Curr. Med. Chem. 2009, 16, 1–20. doi:10.2174/092986709787002826
Return to citation in text: [1] -
Yu, J.-Q.; Shi, Z. C–H Activation; Topics in Current Chemistry, Vol. 292; Springer: Heidelberg, 2010. doi:10.1007/978-3-642-12356-6
Return to citation in text: [1] -
Diederich, F.; Stang, P. J.; Tykwinski, R. R. Acetylene chemistry: chemistry, biology, and material science; Wiley-VCH: Weinheim; Germany, 2005.
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266. doi:10.1021/cr068435d
Return to citation in text: [1] -
Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346. doi:10.1002/anie.200905419
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304. doi:10.1002/anie.201003179
Return to citation in text: [1] [2] [3] [4] -
Brand, J. P.; Waser, J. Org. Lett. 2012, 14, 744. doi:10.1021/ol203289v
Return to citation in text: [1] -
Li, Y.; Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2013, 52, 6743. doi:10.1002/anie.201302210
Return to citation in text: [1] [2] -
Zhdankin, V. V. Curr. Org. Synth. 2005, 2, 121. doi:10.2174/1570179052996982
Return to citation in text: [1] -
Brand, J. P.; Fernández González, D.; Nicolai, S.; Waser, J. Chem. Commun. 2011, 47, 102. doi:10.1039/c0cc02265a
Return to citation in text: [1] -
Zhdankin, V. V.; Kuehl, C. J.; Krasutsky, A. P.; Bolz, J. T.; Simonsen, A. J. J. Org. Chem. 1996, 61, 6547. doi:10.1021/jo960927a
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Synthesis 2012, 44, 1155. doi:10.1055/s-0031-1290589
Return to citation in text: [1] -
Bouma, M. J.; Olofsson, B. Chem.–Eur. J. 2012, 18, 14242. doi:10.1002/chem.201202977
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Chem. Soc. Rev. 2012, 41, 4165. doi:10.1039/c2cs35034c
See for a review on electrophilic alkynylation.
Return to citation in text: [1] -
Wang, Y.; Liu, L.; Zhang, L. Chem. Sci. 2013, 4, 739. doi:10.1039/c2sc21333h
See for a recent example of cooperative gold-zinc catalysis.
Return to citation in text: [1] -
See Supporting Information for a full list of tested Lewis acids.
Return to citation in text: [1] -
No product was obtained with strongly electron-withdrawing substituents, such as cyanide.
Return to citation in text: [1] -
A 5:1 mixture of non-separable products was obtained, which prevented complete assignment of the structure of the major regioisomer.
Return to citation in text: [1] [2] -
The regiochemistry of the alkynylation was determined by 2D NMR experiments after removal of the silyl protecting group on alkyne 10. One other non-identified isomer was observed in the crude mixture by 1H NMR (yield < 5%).
Return to citation in text: [1] -
Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem.–Eur. J. 2012, 18, 5655. doi:10.1002/chem.201200200
Return to citation in text: [1] -
Koller, R.; Stanek, K.; Stolz, D.; Aardoom, R.; Niedermann, K.; Togni, A. Angew. Chem., Int. Ed. 2009, 48, 4332. doi:10.1002/anie.200900974
Return to citation in text: [1] -
See Figure S1 in the Supporting Information File 1.
Return to citation in text: [1]
| 1. | Song, P.-S.; Tapley, K. J., Jr. Photochem. Photobiol. 1979, 29, 1177. doi:10.1111/j.1751-1097.1979.tb07838.x |
| 2. | Gasparro, F. P.; Dallamico, R.; Goldminz, D.; Simmons, E.; Weingold, D. Yale J. Biol. Med. 1989, 62, 579. |
| 3. | Caffieri, S. Photochem. Photobiol. Sci. 2002, 1, 149. doi:10.1039/b107329j |
| 4. | Santana, L.; Uriarte, E.; Roleira, F.; Milhazes, N.; Borges, F. Curr. Med. Chem. 2004, 11, 3239. doi:10.2174/0929867043363721 |
| 9. | Cacchi, S.; Fabrizi, G.; Goggiomani, A. Heterocycles 2002, 56, 613. doi:10.3987/REV-01-SR(K)1 |
| 10. | Hou, X.-L.; Yang, Z.; Yeung, K.-S.; Wong, H. N. C. Five-membered ring systems: furans and benzofurans; Progress in Heterocyclic Chemistry, Vol. 18; Elsevier, 2007; pp 187–217. doi:10.1016/S0959-6380(07)80011-0 |
| 11. | De Luca, L.; Nieddu, G.; Porcheddu, A.; Giacomelli, G. Curr. Med. Chem. 2009, 16, 1–20. doi:10.2174/092986709787002826 |
| 24. | Zhdankin, V. V.; Kuehl, C. J.; Krasutsky, A. P.; Bolz, J. T.; Simonsen, A. J. J. Org. Chem. 1996, 61, 6547. doi:10.1021/jo960927a |
| 25. | Brand, J. P.; Waser, J. Synthesis 2012, 44, 1155. doi:10.1055/s-0031-1290589 |
| 26. | Bouma, M. J.; Olofsson, B. Chem.–Eur. J. 2012, 18, 14242. doi:10.1002/chem.201202977 |
| 27. |
Brand, J. P.; Waser, J. Chem. Soc. Rev. 2012, 41, 4165. doi:10.1039/c2cs35034c
See for a review on electrophilic alkynylation. |
| 8. | Mitsui, C.; Soeda, J.; Miwa, K.; Tsuji, H.; Takeya, J.; Nakamura, E. J. Am. Chem. Soc. 2012, 134, 5448. doi:10.1021/ja2120635 |
| 19. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304. doi:10.1002/anie.201003179 |
| 28. |
Wang, Y.; Liu, L.; Zhang, L. Chem. Sci. 2013, 4, 739. doi:10.1039/c2sc21333h
See for a recent example of cooperative gold-zinc catalysis. |
| 6. | Mason, J. W. N. Engl. J. Med. 1987, 316, 455. doi:10.1056/NEJM198702193160807 |
| 7. | Bogazzi, F.; Tomisti, L.; Bartalena, L.; Aghini-Lombardi, F.; Martino, E. J. Endocrinol. Invest. 2012, 35, 340. doi:10.3275/8298 |
| 22. | Zhdankin, V. V. Curr. Org. Synth. 2005, 2, 121. doi:10.2174/1570179052996982 |
| 23. | Brand, J. P.; Fernández González, D.; Nicolai, S.; Waser, J. Chem. Commun. 2011, 47, 102. doi:10.1039/c0cc02265a |
| 5. | Bickoff, E. M.; Booth, A. N.; Lyman, R. L.; Livingston, A. L.; Thompson, C. R.; Deeds, F. Science 1957, 126, 969. doi:10.1126/science.126.3280.969-a |
| 21. | Li, Y.; Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2013, 52, 6743. doi:10.1002/anie.201302210 |
| 18. | Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346. doi:10.1002/anie.200905419 |
| 14. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335 |
| 15. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x |
| 16. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g |
| 17. | Arcadi, A. Chem. Rev. 2008, 108, 3266. doi:10.1021/cr068435d |
| 21. | Li, Y.; Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2013, 52, 6743. doi:10.1002/anie.201302210 |
| 13. | Diederich, F.; Stang, P. J.; Tykwinski, R. R. Acetylene chemistry: chemistry, biology, and material science; Wiley-VCH: Weinheim; Germany, 2005. |
| 12. | Yu, J.-Q.; Shi, Z. C–H Activation; Topics in Current Chemistry, Vol. 292; Springer: Heidelberg, 2010. doi:10.1007/978-3-642-12356-6 |
| 19. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304. doi:10.1002/anie.201003179 |
| 30. | No product was obtained with strongly electron-withdrawing substituents, such as cyanide. |
| 19. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304. doi:10.1002/anie.201003179 |
| 19. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304. doi:10.1002/anie.201003179 |
| 34. | Koller, R.; Stanek, K.; Stolz, D.; Aardoom, R.; Niedermann, K.; Togni, A. Angew. Chem., Int. Ed. 2009, 48, 4332. doi:10.1002/anie.200900974 |
| 32. | The regiochemistry of the alkynylation was determined by 2D NMR experiments after removal of the silyl protecting group on alkyne 10. One other non-identified isomer was observed in the crude mixture by 1H NMR (yield < 5%). |
| 33. | Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem.–Eur. J. 2012, 18, 5655. doi:10.1002/chem.201200200 |
| 31. | A 5:1 mixture of non-separable products was obtained, which prevented complete assignment of the structure of the major regioisomer. |
| 31. | A 5:1 mixture of non-separable products was obtained, which prevented complete assignment of the structure of the major regioisomer. |
© 2013 Li and Waser; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)