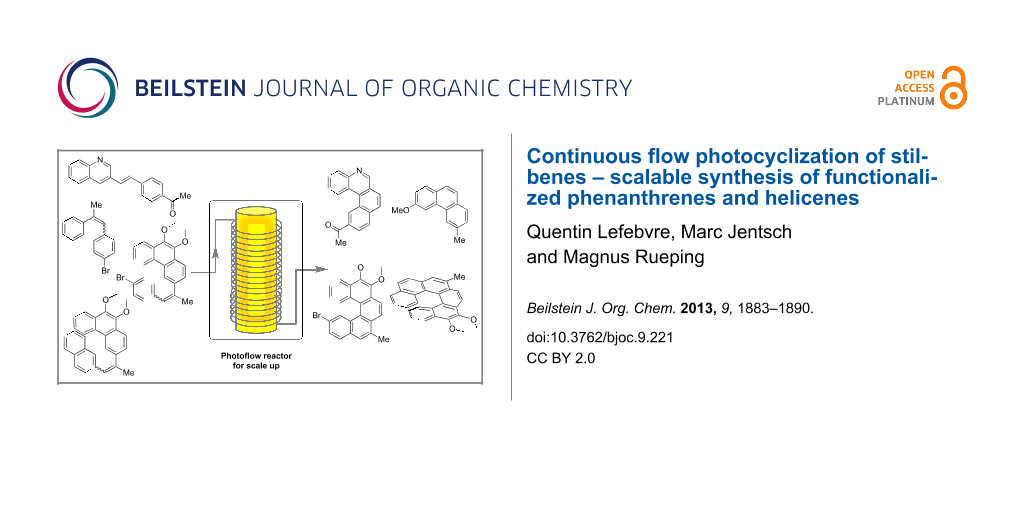
Abstract
A continuous flow oxidative photocyclization of stilbene derivatives has been developed which allows the scalable synthesis of backbone functionalized phenanthrenes and helicenes of various sizes in good yields.

Graphical Abstract
Introduction
Phenanthrenes are versatile intermediates toward polycyclic aromatic hydrocarbons which are relevant for materials sciences, as well as toward helicenes, an intriguing class of molecules which show remarkable chiroptical properties due to their helical pitch. The rapidly expanding field of application of helicene-like molecules in materials sciences and optics demands the development of scalable and flexible syntheses [1-5].
Following the pioneering examples of Scholz [6] and Martin [7] in 1967, the photocyclization of stilbene derivatives under UV-light irradiation is now a classical method for the synthesis of phenanthrenes and helicene-like molecules [8]. However, the scalability of these reactions is limited by the required low concentration (~10−3 mol·L−1) and usually long reaction times (>20 h). Therefore, most of the applications of this method are limited to small scale (<0.5 mmol), making it unsuitable for gram-scale synthesis of helicene-like molecules [9-12]. Much effort has devoted to the development of alternative pathways toward helicenes, but most approaches require lengthy syntheses of the precursors [13-15]. It should be noted that an elegant semi-one-pot procedure for the synthesis of phenanthrenes from styrenes and benzene was recently reported [16]. However, also in this case, the scale of the reaction is limited by the size of the photoreactor.
Reactions in flow are typically faster and cleaner compared to the corresponding reactions in batch. A flow setup is particularly well-suited for photo-catalysed reactions as the efficiency of the transformation is no longer related to the scale [17-21]. Therefore, the development of an efficient protocol for the photocyclization of stilbene derivatives in flow would be of great interest. A recent contribution described the flow-synthesis of [5]helicene under visible light in the presence of a sensitizer [22], but, to the best of our knowledge, no reports on broadly applicable light-induced oxidative photocyclizations in flow are known. Herein, we report the first photocyclization of polysubstituted olefins using a continuous flow process and discuss advantages and limitations of this new protocol. Generally, the oxidative photodehydrogenation of E-stilbene results in the formation of phenanthrene (Scheme 1). In this reaction the E-olefin (or a E/Z mixture of the olefin) is photoisomerised to the reactive Z-olefin, which undergoes photocyclization. The corresponding dihydrophenanthene is subsequently oxidized with iodine to give the desired phenanthrene and HI, which can be quenched by propylene oxide or THF.
![[1860-5397-9-221-i1]](/bjoc/content/inline/1860-5397-9-221-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photocyclization of stilbene to phenanthrene.
Scheme 1: Photocyclization of stilbene to phenanthrene.
Results and Discussion
In order to accomplish a first general photocyclization we started with the photocyclization of stilbene. Optimization of the reaction conditions in a small flow setup (5 mL FEP tubing, 150 W UV-lamp) is presented in Table 1. Although both THF and propylene oxide showed good ability to quench HI, we choose THF as additive due to its lower cost, volatility and toxicity [23,24]. Under the optimized conditions, E-stilbene (1a) could be converted to phenanthrene (2a) in 95% NMR yield with a retention time of 83 min (Table 1, entry 5).
Table 1: Proof of principle and screening of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-221-i4.svg?max-width=637&scale=1.0)
|
||||
| Entrya |
Additive
(20 equiv) |
Concentration
(mol/L) |
Flow rate
(mL/min) |
Yieldb (%) |
|---|---|---|---|---|
| 1 | propylene oxide | 0.01 | 0.06 | 33 |
| 2 | THF | 0.01 | 0.04 | 37 |
| 3 | THF | 0.01 | 0.02 | 44 |
| 4 | propylene oxide | 0.001 | 0.06 | 99 |
| 5 | THF | 0.002 | 0.06 | 95 |
| 6 | cyclohexene | 0.002 | 0.06 | –c |
| 7 | THF | 0.002 | 0.08 | 68 |
| 8 | THF | 0.003 | 0.06 | 50 |
a1.1 equiv of iodine were used. The solvent was dry toluene. bDetermined by 1H NMR using mesitylene as internal standard. cMainly Z-stilbene was observed.
The flow-reactor setup used for the optimization (Table 1) is shown in Figure 1. UV-transparent ethylene propylene copolymer capillary (FEP, outer/inner diameter 1/0.5 mm, total volume 5 mL) was tightly wrapped around the water-cooling unit (Duran glass) of a high-pressure mercury lamp (TQ 150, UV-Consulting Peschl). Further optimization and scope was performed with a similar setup using a bigger capillary (FEP, outer/inner diameter 4/2 mm, total volume 24 mL). For the sake of safety and to enhance the efficiency of irradiation by reflection, the setup was placed into a laboratory Dewar flask and the exposed parts of the setup were covered with aluminium foil (the temperature inside the Dewar was determined to be around 40 °C). The reaction mixture was injected into the system using a syringe pump and collected at the outlet of the tubing into a round bottom flask.
![[1860-5397-9-221-1]](/bjoc/content/figures/1860-5397-9-221-1.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Flow-reactor setup used in the optimization study.
Figure 1: Flow-reactor setup used in the optimization study.
Additionally, we developed a 5-fold bigger setup to enhance the throughput (Table 2). Slightly longer retention times were required, and degassed toluene provided better yields. Finally, phenanthrene (2a) was obtained in 94% yield when a flow rate of 0.2 mL/min (retention time of 120 min) was applied (Table 2, entry 4).
Table 2: Optimisation of the scale-up setup.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-221-i5.svg?max-width=637&scale=1.0)
|
|||
| Entrya |
Additive
(20 equiv) |
Flow rate
(mL/min) |
Yieldb
(%) |
|---|---|---|---|
| 1 | propylene oxide | 0.2 | 66 |
| 2 | THF | 0.25 | 40 |
| 3 | THF | 0.2 | 85 |
| 4c | THF | 0.2 | 94 |
a1.1 equiv of iodine were used. Dry toluene was used as solvent. bDetermined by 1H NMR using mesitylene as internal standard. cDegassed toluene was used.
With the optimised conditions in hand, we explored the scope of the reaction. Although photocyclization of disubstituted olefins in batch was well documented in the literature, only few cases of photocyclization of tri- and tetrasubstituted olefins were reported [16,25]. Therefore, we decided to demonstrate our methodology on both di- and trisubstituted olefins (Table 3). We disclose here the first photocyclization of trisubstituted olefins in flow, giving access to backbone-functionalised phenanthrene derivatives.
Table 3: Scope of the photocyclization of stilbene derivatives in continuous flow to give substituted phenanthrenes.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-221-i6.svg?max-width=637&scale=1.0)
|
|||
| Entrya | Substrate | Product (2) | Yieldb (%) |
|---|---|---|---|
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-221-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-221-i8.svg?max-width=637&scale=1.0)
2a |
64 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-221-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-221-i10.svg?max-width=637&scale=1.0)
2b |
64 |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-221-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-221-i12.svg?max-width=637&scale=1.0)
2c |
61 |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-221-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-221-i14.svg?max-width=637&scale=1.0)
2d |
77 |
| 5 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-221-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-221-i16.svg?max-width=637&scale=1.0)
2e and 2e’ |
31/34 |
| 6 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-221-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-221-i18.svg?max-width=637&scale=1.0)
2f |
77 |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-221-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-221-i20.svg?max-width=637&scale=1.0)
2g |
89 |
| 8 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-221-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-221-i22.svg?max-width=637&scale=1.0)
2h |
64 |
aReaction conditions: 1.1 equiv iodine, 20 equiv THF, UV-light, 2 h retention time. bYield after chromatography.
We choose stilbenes with bromide and methyl substituents, as the latter can be used in subsequent oxidation, deprotonation, and radical addition reactions, whereas the former opens access to various functional groups via lithium-halogen exchange or cross-coupling chemistry. Methoxy groups were also tolerated, but nitro- and amino groups containing stilbenes showed low conversion or decomposition. Meta-substituted substrates gave inseparable regioisomers, and ortho-substitution led to low conversion. In the case of substrate 1e, a separable 1:1 mixture of regioisomers 2e and 2e’ was obtained (Table 3, entry 5). However, generally a series of stilbenes reacted smoothly to the desired phenantrenes in good yields.
Recently, an unexpected synthesis of [4]helicenes was disclosed [26]. However, no 2-substituted [4]helicenes were synthesised using this method. Therefore, we investigated the photocyclization of the corresponding stilbene derivatives in our flow setup, for both di- and trisubstituted olefins (Table 4).
Table 4: Scope: synthesis of [4]helicenes by photocyclization in flow.
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-221-i23.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Substrate | Product (2) | Yieldb (%) | |
|---|---|---|---|---|
| 1 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-221-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-221-i25.svg?max-width=637&scale=1.0)
2i |
85 | |
| 2 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-221-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-221-i27.svg?max-width=637&scale=1.0)
2j |
75 | |
| 3 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-221-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-221-i29.svg?max-width=637&scale=1.0)
2k |
74 | |
| 4c |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-221-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-221-i31.svg?max-width=637&scale=1.0)
2k and 2k’ |
40/41 | |
| 5 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-221-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-221-i33.svg?max-width=637&scale=1.0)
2l |
75 | |
| 6 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-221-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-221-i35.svg?max-width=637&scale=1.0)
2m |
73 | |
| 7 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-221-i36.svg?max-width=637&scale=1.0)
|
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-221-i37.svg?max-width=637&scale=1.0)
2n |
99 | |
aReaction conditions: 1.1 equiv iodine, 20 equiv THF, UV-light, 2 h retention time. bYield after chromatography. cReaction was performed in dry, degassed acetonitrile.
Again, various functional groups were tolerated in the flow photocyclization. Interestingly, if substrate 1k was irradiated in toluene a single regioisomer 2k was isolated (Table 4, entry 3), whereas the reaction in acetonitrile resulted in a separable 1:1 mixture of regioisomers 2k and 2k’ (Table 4, entry 4).
Finally, we decided to apply our photo-flow methodology in the synthesis of functionalised [5]helicenes and [6]helicenes. We identified 3-acetyl-9,10-dimethoxyphenanthrene [27] as a powerful intermediate for the two-step synthesis of functionalisable helically chiral products. As shown in Scheme 2, Wittig or Horner–Wadsworth–Emmons reactions gave the corresponding olefins 1o–r in good yields, which were subjected to photocyclization in flow using the optimised conditions for the simpler stilbenes. In the [5]helicene series, in each case only one product was isolated in moderate to good yield. The bromoolefin 1o gave exclusively the desired helicene 2o, but in the case of methyl- and methoxyolefins 1p and 1q, only the corresponding benzo[ghi]perylenes 2p and 2q were observed. Benzo[ghi]perylenes are typical byproducts observed in the photocyclization of [5]helicene-like molecules. Reactions to obtain selectively helicenes or benzo[ghi]perylenes, regardless of the substitution pattern, are still a challenging task. A functionalizable [6]helicene (2r) was obtained along with its ribbon-like regioisomer 2r’ in a 1:1 ratio and 75% combined yield.
![[1860-5397-9-221-i2]](/bjoc/content/inline/1860-5397-9-221-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Photo-flow synthesis of [5]- and [6]helicenes. aFor experimental details see Supporting Information File 1. bReaction conditions: 1.1 equiv iodine, 20 equiv THF, UV-light, 2 h retention time.
Scheme 2: Photo-flow synthesis of [5]- and [6]helicenes. aFor experimental details see Supporting Information File 1. bReaction conditions...
In order to demonstrate the utility of the flow process we decided to scale up the synthesis of helical compound 2o. To our delight, we observed that the [5]helicene precursor 1o underwent photocyclization with considerably shorter retention times compared to the standard stilbene derivatives (Scheme 3). Thus, we prepared the [5]helicene derivative 2o with up to 60 mg/h (for the full optimisation table, see Supporting Information File 1).
![[1860-5397-9-221-i3]](/bjoc/content/inline/1860-5397-9-221-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scale up synthesis of the [5]helicene derivative 2o.
Scheme 3: Scale up synthesis of the [5]helicene derivative 2o.
Conclusion
In summary, we have developed a new photo-flow methodology [28-32] for the synthesis of phenanthrenes and helicenes. Although photocyclization of stilbene derivatives was disclosed more than 40 years ago, this is the first report of UV-light-driven photocyclization in flow. In general phenantrenes as well as [4]-, [5]- and [6]helicenes with different substitution patterns are obtained in good to excellent yields. In addition our first attempts to scale up the flow photocyclization reactions were successful providing the opportunity for multi-gram syntheses.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2013, 42, 968–1006. doi:10.1039/c2cs35154d
Return to citation in text: [1] -
Gingras, M.; Félix, G.; Peresutti, R. Chem. Soc. Rev. 2013, 42, 1007–1050. doi:10.1039/c2cs35111k
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2013, 42, 1051–1095. doi:10.1039/c2cs35134j
Return to citation in text: [1] -
Moorthy, J. N.; Mandal, S.; Mukhopadhyay, A.; Samanta, S. J. Am. Chem. Soc. 2013, 135, 6872–6884. doi:10.1021/ja312027c
See for a recent example of application of a carbohelicene in optics.
Return to citation in text: [1] -
Scholz, M.; Mühlstädt, M.; Dietz, F. Tetrahedron Lett. 1967, 8, 665–668. doi:10.1016/S0040-4039(00)90569-0
Return to citation in text: [1] -
Flammang-Barbieux, M.; Nasielski, J.; Martin, R. H. Tetrahedron Lett. 1967, 8, 743–744. doi:10.1016/S0040-4039(00)90586-0
Return to citation in text: [1] -
Liu, L.; Yang, B.; Katz, T. J.; Poindexter, M. K. J. Org. Chem. 1991, 56, 3769–3775. doi:10.1021/jo00012a005
Return to citation in text: [1] -
Terfort, A.; Görls, H.; Brunner, H. Synthesis 1997, 79–86. doi:10.1055/s-1997-1498
Return to citation in text: [1] -
Reetz, M. T.; Sostmann, S. J. Organomet. Chem. 2000, 603, 105–109. doi:10.1016/S0022-328X(00)00173-X
Return to citation in text: [1] -
El Abed, R.; Ben Hassine, B.; Genêt, J.-P.; Gorsane, M.; Marinetti, A. Eur. J. Org. Chem. 2004, 1517–1522. doi:10.1002/ejoc.200300470
Return to citation in text: [1] -
El Abed, R.; Aloui, F.; Genêt, J.-P.; Ben Hassine, B.; Marinetti, A. J. Organomet. Chem. 2007, 692, 1156–1160. doi:10.1016/j.jorganchem.2006.11.022
Return to citation in text: [1] -
Stará, I. G.; Alexandrová, Z.; Teplý, F.; Sehnal, P.; Starý, I.; Šaman, D.; Buděšínský, M.; Cvaèka, J. Org. Lett. 2005, 7, 2547–2550. doi:10.1021/ol047311p
Return to citation in text: [1] -
Míšek, J.; Teplý, F.; Stará, I. G.; Tichý, M.; Šaman, D.; Císařová, I.; Vojtíšek, P.; Starý, I. Angew. Chem. 2008, 120, 3232–3235. doi:10.1002/ange.200705463
Angew. Chem., Int. Ed. 2008, 47, 3188–3191. doi:10.1002/anie.200705463
Return to citation in text: [1] -
Weimar, M.; Correa da Costa, R.; Lee, F.-H.; Fuchter, M. J. Org. Lett. 2013, 15, 1706–1709. doi:10.1021/ol400493x
Return to citation in text: [1] -
Li, H.; He, K.-H.; Liu, J.; Wang, B.-Q.; Zhao, K.-Q.; Hu, P.; Shi, Z.-J. Chem. Commun. 2012, 48, 7028–7030. doi:10.1039/c2cc33100d
Return to citation in text: [1] [2] -
Matsushita, Y.; Ichimura, T.; Ohba, N.; Kumada, S.; Sakeda, K.; Suzuki, T.; Tanibata, H.; Murata, T. Pure Appl. Chem. 2007, 79, 1959–1968. doi:10.1351/pac200779111959
Return to citation in text: [1] -
Oelgemöller, M.; Shvydkiv, O. Molecules 2011, 16, 7522–7550. doi:10.3390/molecules16097522
Return to citation in text: [1] -
Knowles, J. P.; Elliott, L. D.; Booker-Milburn, K. I. Beilstein J. Org. Chem. 2012, 8, 2025–2052. doi:10.3762/bjoc.8.229
Return to citation in text: [1] -
Oelgemöller, M. Chem. Eng. Technol. 2012, 35, 1144–1152. doi:10.1002/ceat.201200009
Return to citation in text: [1] -
Shvydkiv, O.; Oelgemöller, M. Microphotochemistry: Photochemical Synthesis in Microstructed Flow Reactors. In CRC Handbook of Organic Photochemistry and Photobiology; Griesbeck, A.; Oelgemöller, M.; Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp 125–178.
Return to citation in text: [1] -
Hernandez-Perez, A. C.; Vlassova, A.; Collins, S. K. Org. Lett. 2012, 14, 2988–2991. doi:10.1021/ol300983b
Return to citation in text: [1] -
Talele, H. R.; Gohil, M. J.; Bedekar, A. V. Bull. Chem. Soc. Jpn. 2009, 82, 1182–1186. doi:10.1246/bcsj.82.1182
Return to citation in text: [1] -
Talele, H. R.; Chaudhary, A. R.; Patel, P. R.; Bedekar, A. V. ARKIVOC 2011, No. ix, 15–37. doi:10.3998/ark.5550190.0012.902
Return to citation in text: [1] -
Yavari, K.; Moussa, S.; Ben Hassine, B.; Retailleau, P.; Voituriez, A.; Marinetti, A. Angew. Chem. 2012, 124, 6852–6856. doi:10.1002/anie.201202024
Angew. Chem., Int. Ed. 2012, 51, 6748–6752 doi:10.1002/anie.201202024
Return to citation in text: [1] -
Truong, T.; Daugulis, O. Chem. Sci. 2012, 4, 531–535. doi:10.1039/c2sc21288a
Return to citation in text: [1] -
Wang, D. Z.; Katz, T. J. J. Org. Chem. 2005, 70, 8497–8502. doi:10.1021/jo0512913
Return to citation in text: [1] -
Liao, H.-H.; Hsiao, C.-C.; Sugiono, E.; Rueping, M. Chem. Commun. 2013, 49, 7953–7955. doi:10.1039/c3cc43996h
See for recent applications of flow chemistry from our group.
Return to citation in text: [1] -
Rueping, M.; Vila, C.; Bootwicha, T. ACS Catal. 2013, 3, 1676–1680. doi:10.1021/cs400350j
Return to citation in text: [1] -
Rueping, M.; Bootwicha, T.; Sugiono, E. Beilstein J. Org. Chem. 2012, 8, 300–307. doi:10.3762/bjoc.8.32
Return to citation in text: [1] -
Rueping, M.; Bootwicha, T.; Baars, H.; Sugiono, E. Beilstein J. Org. Chem. 2011, 7, 1680–1687. doi:10.3762/bjoc.7.198
Return to citation in text: [1] -
Rueping, M.; Bootwicha, T.; Sugiono, E. Adv. Synth. Catal. 2010, 352, 2961–2965. doi:10.1002/adsc.201000538
Return to citation in text: [1]
| 1. | Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r |
| 2. | Gingras, M. Chem. Soc. Rev. 2013, 42, 968–1006. doi:10.1039/c2cs35154d |
| 3. | Gingras, M.; Félix, G.; Peresutti, R. Chem. Soc. Rev. 2013, 42, 1007–1050. doi:10.1039/c2cs35111k |
| 4. | Gingras, M. Chem. Soc. Rev. 2013, 42, 1051–1095. doi:10.1039/c2cs35134j |
| 5. |
Moorthy, J. N.; Mandal, S.; Mukhopadhyay, A.; Samanta, S. J. Am. Chem. Soc. 2013, 135, 6872–6884. doi:10.1021/ja312027c
See for a recent example of application of a carbohelicene in optics. |
| 9. | Terfort, A.; Görls, H.; Brunner, H. Synthesis 1997, 79–86. doi:10.1055/s-1997-1498 |
| 10. | Reetz, M. T.; Sostmann, S. J. Organomet. Chem. 2000, 603, 105–109. doi:10.1016/S0022-328X(00)00173-X |
| 11. | El Abed, R.; Ben Hassine, B.; Genêt, J.-P.; Gorsane, M.; Marinetti, A. Eur. J. Org. Chem. 2004, 1517–1522. doi:10.1002/ejoc.200300470 |
| 12. | El Abed, R.; Aloui, F.; Genêt, J.-P.; Ben Hassine, B.; Marinetti, A. J. Organomet. Chem. 2007, 692, 1156–1160. doi:10.1016/j.jorganchem.2006.11.022 |
| 8. | Liu, L.; Yang, B.; Katz, T. J.; Poindexter, M. K. J. Org. Chem. 1991, 56, 3769–3775. doi:10.1021/jo00012a005 |
| 7. | Flammang-Barbieux, M.; Nasielski, J.; Martin, R. H. Tetrahedron Lett. 1967, 8, 743–744. doi:10.1016/S0040-4039(00)90586-0 |
| 27. | Wang, D. Z.; Katz, T. J. J. Org. Chem. 2005, 70, 8497–8502. doi:10.1021/jo0512913 |
| 6. | Scholz, M.; Mühlstädt, M.; Dietz, F. Tetrahedron Lett. 1967, 8, 665–668. doi:10.1016/S0040-4039(00)90569-0 |
| 28. |
Liao, H.-H.; Hsiao, C.-C.; Sugiono, E.; Rueping, M. Chem. Commun. 2013, 49, 7953–7955. doi:10.1039/c3cc43996h
See for recent applications of flow chemistry from our group. |
| 29. | Rueping, M.; Vila, C.; Bootwicha, T. ACS Catal. 2013, 3, 1676–1680. doi:10.1021/cs400350j |
| 30. | Rueping, M.; Bootwicha, T.; Sugiono, E. Beilstein J. Org. Chem. 2012, 8, 300–307. doi:10.3762/bjoc.8.32 |
| 31. | Rueping, M.; Bootwicha, T.; Baars, H.; Sugiono, E. Beilstein J. Org. Chem. 2011, 7, 1680–1687. doi:10.3762/bjoc.7.198 |
| 32. | Rueping, M.; Bootwicha, T.; Sugiono, E. Adv. Synth. Catal. 2010, 352, 2961–2965. doi:10.1002/adsc.201000538 |
| 22. | Hernandez-Perez, A. C.; Vlassova, A.; Collins, S. K. Org. Lett. 2012, 14, 2988–2991. doi:10.1021/ol300983b |
| 16. | Li, H.; He, K.-H.; Liu, J.; Wang, B.-Q.; Zhao, K.-Q.; Hu, P.; Shi, Z.-J. Chem. Commun. 2012, 48, 7028–7030. doi:10.1039/c2cc33100d |
| 25. |
Yavari, K.; Moussa, S.; Ben Hassine, B.; Retailleau, P.; Voituriez, A.; Marinetti, A. Angew. Chem. 2012, 124, 6852–6856. doi:10.1002/anie.201202024
Angew. Chem., Int. Ed. 2012, 51, 6748–6752 doi:10.1002/anie.201202024 |
| 17. | Matsushita, Y.; Ichimura, T.; Ohba, N.; Kumada, S.; Sakeda, K.; Suzuki, T.; Tanibata, H.; Murata, T. Pure Appl. Chem. 2007, 79, 1959–1968. doi:10.1351/pac200779111959 |
| 18. | Oelgemöller, M.; Shvydkiv, O. Molecules 2011, 16, 7522–7550. doi:10.3390/molecules16097522 |
| 19. | Knowles, J. P.; Elliott, L. D.; Booker-Milburn, K. I. Beilstein J. Org. Chem. 2012, 8, 2025–2052. doi:10.3762/bjoc.8.229 |
| 20. | Oelgemöller, M. Chem. Eng. Technol. 2012, 35, 1144–1152. doi:10.1002/ceat.201200009 |
| 21. | Shvydkiv, O.; Oelgemöller, M. Microphotochemistry: Photochemical Synthesis in Microstructed Flow Reactors. In CRC Handbook of Organic Photochemistry and Photobiology; Griesbeck, A.; Oelgemöller, M.; Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp 125–178. |
| 16. | Li, H.; He, K.-H.; Liu, J.; Wang, B.-Q.; Zhao, K.-Q.; Hu, P.; Shi, Z.-J. Chem. Commun. 2012, 48, 7028–7030. doi:10.1039/c2cc33100d |
| 13. | Stará, I. G.; Alexandrová, Z.; Teplý, F.; Sehnal, P.; Starý, I.; Šaman, D.; Buděšínský, M.; Cvaèka, J. Org. Lett. 2005, 7, 2547–2550. doi:10.1021/ol047311p |
| 14. |
Míšek, J.; Teplý, F.; Stará, I. G.; Tichý, M.; Šaman, D.; Císařová, I.; Vojtíšek, P.; Starý, I. Angew. Chem. 2008, 120, 3232–3235. doi:10.1002/ange.200705463
Angew. Chem., Int. Ed. 2008, 47, 3188–3191. doi:10.1002/anie.200705463 |
| 15. | Weimar, M.; Correa da Costa, R.; Lee, F.-H.; Fuchter, M. J. Org. Lett. 2013, 15, 1706–1709. doi:10.1021/ol400493x |
| 23. | Talele, H. R.; Gohil, M. J.; Bedekar, A. V. Bull. Chem. Soc. Jpn. 2009, 82, 1182–1186. doi:10.1246/bcsj.82.1182 |
| 24. | Talele, H. R.; Chaudhary, A. R.; Patel, P. R.; Bedekar, A. V. ARKIVOC 2011, No. ix, 15–37. doi:10.3998/ark.5550190.0012.902 |
© 2013 Lefebvre et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)