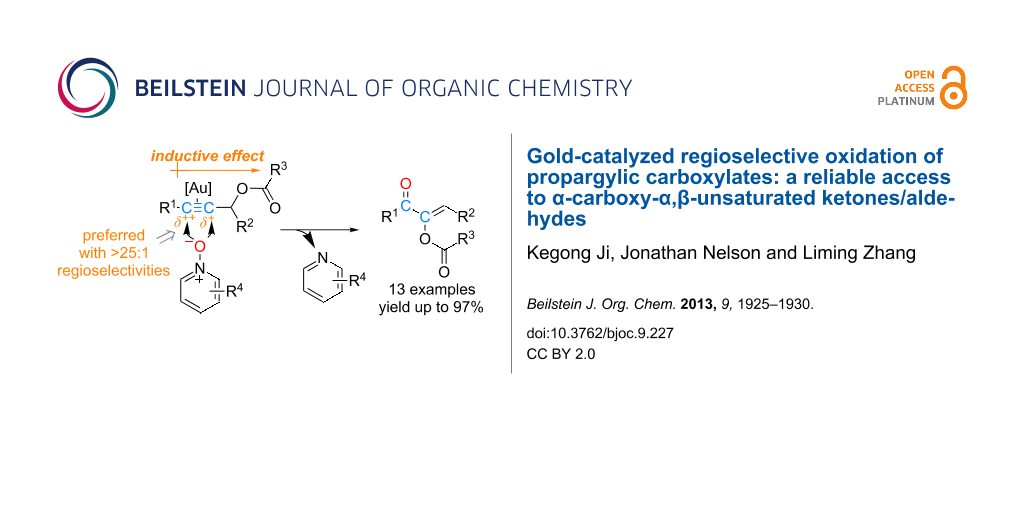
Abstract
Gold-catalyzed intermolecular oxidation of carboxylates of primary or secondary propargylic alcohols are realized with excellent regioselectivity, which is ascribed to inductive polarization of the C–C triple bond by the electron-withdrawing carboxy group. The gold carbene intermediates thus generated undergo selective 1,2-acyloxy migration over a 1,2-C–H insertion, and the selectivities could be dramatically improved by the use of a P,S-bidentate ligand, which is proposed to enable the formation of tris-coordinated and hence less electrophilic gold carbene species. α-Carboxy α,β-unsaturated ketones/aldehydes can be obtained with fair to excellent yields.

Graphical Abstract
Introduction
We reported in 2010 [1] that α-oxo gold carbenes could be conveniently generated as reactive intermediates in gold-catalyzed intermolecular oxidation of alkynes. By using pyridine N-oxides [1] and later 8-substituted quinoline N-oxides [2] as the external oxidants, this approach permits a safe and efficient access to α-oxo gold carbenes without resorting to the dediazotization strategy [3-5] using hazardous and potentially explosive diazo substrates (Scheme 1). Since then an array of versatile synthetic methods has been developed based on the general approach by us [2,6-12] and other researchers [13-20], thus making it an exciting area for further advancing gold chemistry.
![[1860-5397-9-227-i1]](/bjoc/content/inline/1860-5397-9-227-i1.png?scale=1.6&max-width=1024&background=FFFFFF)
Scheme 1: Generation of α-oxo gold carbenes via intermolecular oxidation of alkynes: a non-diazo approach.
Scheme 1: Generation of α-oxo gold carbenes via intermolecular oxidation of alkynes: a non-diazo approach.
Among various types of alkynes examined, internal alkynes, while without incident in the generation of the gold carbene intermediates, present an additional challenge, namely how to control the regioselectivity of the oxidation. We reported previously that synthetically useful regioselectivity could be achieved if the two ends of the C–C triple bond are biased by a steric bulk and/or via conjugation (Scheme 2). In our continued effort to reveal regioselectivities of this oxidation with different types of internal alkynes, we examined propargylic carboxylates, which have served as a versatile platform for the development of a diverse range of gold catalysis [21]. Herein we report our findings and the development of a reliable synthesis of α-carboxy α,β-unsaturated ketones/aldehydes.
![[1860-5397-9-227-i2]](/bjoc/content/inline/1860-5397-9-227-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Gold-catalyzed regioselective oxidation of a sterically biased internal alkyne.
Scheme 2: Gold-catalyzed regioselective oxidation of a sterically biased internal alkyne.
Results and Discussion
We began by subjecting the propargylic acetate 4a to the suitable conditions developed in our previous study, namely IPrAuNTf2 (5 mol %) and 8-methylquinoline N-oxide (3, 1.5 equiv) in 1,2-dichloroethane at ambient temperature. To our delight, the reaction proceeded efficiently, yielding the α-acetoxyenone 5a-OAc (Z/E >50:1) and the isomeric β-acetoxyenone 5a-H in an excellent combined 92% yield along with a minute amount of the enone 6 (<0.5%, Scheme 3); moreover, 5a-OAc is favored over 5a-H by a ratio of ~7:1. Of particular importance is that the anticipated isomer 5a’, accessible via the gold carbene B’ from a regioisomeric alkyne oxidation, was not positively detected due to the trace amount (<0.5%), thereby revealing an exceptional level of regioselectivity in the oxidation of this type of internal alkynes. The formations of 5a-OAc and 5a-H are rationalized as the results of divergent transformations of the α-oxo gold carbene B: the former is formed via a two-step 2,3-acetoxy migration [22,23], and the latter most likely via a concerted carbene 1,2-C–H insertion[2]. The selective formation of the Z isomer of 5a-OAc can be attributed to that B adopts a preferred conformation, as detailed in Scheme 3, in order to avoid steric interaction between Me and the acyl moiety. It needs to be noted that a related intramolecular version of this reaction has been previously reported [24].
![[1860-5397-9-227-i3]](/bjoc/content/inline/1860-5397-9-227-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Gold-catalyzed oxidation of the propargylic acetate 4a and the mechanistic rationale.
Scheme 3: Gold-catalyzed oxidation of the propargylic acetate 4a and the mechanistic rationale.
An alternative mechanism for the formation of 5a-OAc is also shown in Scheme 3 (the top half). Instead of initially undergoing oxidative gold carbene formation, a gold-promoted 1,2-acetoxy migration [25] would generate a vinyl gold carbene intermediate (i.e., C), which can then be oxidized by 3 to yield the product. However, this scenario is deemed unlikely by the following observations and considerations: a) propargylic carboxylates of type 4a with an internal C–C triple bond typically undergo facile 3,3-rearrangements [26-30] instead of 1,2-acyloxy migrations. The former process would eventually lead to the formation of the enones 6 [31]. Due to hydrolysis, only under thermal and anhydrous conditions products derived from the latter processes can be predominantly formed [32]; under our conditions (at ambient temperature and without exclusion of moisture), the enone 6 was indeed detected but in a minute amount, suggesting that the 1,2-acyloxy migration might be an even less meaningful event in the reaction; b) it is known that the gold carbenes of type B can be readily oxidized by Ph2S=O [33], which, however, is an inefficient oxidant for generating α-oxo gold carbenes of type A via alkyne oxidation [34,35]; when the N-oxide 3 is replaced by the sulfoxide, 5a-OAc was formed in only 5% yield even at 60 °C after 12 h (Scheme 4); moreover, the major product in the reaction was the expected enone 6 (56% yield, 88% conversion) due to a dominant gold-catalyzed 3,3-rearrangement; c) this alternative could not rationalize the formation of 5a-H.
![[1860-5397-9-227-i4]](/bjoc/content/inline/1860-5397-9-227-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: A drastically different outcome by using diphenyl sulfoxide as the oxidant.
Scheme 4: A drastically different outcome by using diphenyl sulfoxide as the oxidant.
The fact that in the presence of the oxidant 3 the previously observed facile transformations of propargylic carboxylates (i.e., 3,3-rearrangement and 1,2-acyloxy migration) are no longer competitive with the oxidative catalysis is surprising and suggests that this oxidation process could divert substrates from other well established, facile gold catalysis to the formation of distinctively different functional products in the presence of oxidants.
The relatively low selectivity (i.e., ~7:1) of 5a-OAc over 5a-H was drastically improved upon catalyst screening. It was eventually found that the ratio could reach >200:1 by using the gold(I) catalyst derived from our previously developed bulky P,S-bidentate ligand L1 (Figure 1) [11]. A similarly high selectivity was also achieved by using the P,N-bidentate ligand Mor-DalPhos [36,37]. However, the Z/E ratios of 5a-OAc in the former case is ~13:1, better than ~7:1 in the latter case, albeit both lower than that by IPrAuNTf2 (>50:1, see Scheme 3). The enhanced preference of AcO migration en route to the formation of 5a-OAc over the 1,2-C-H insertion is attributed to attenuation of the electrophilicity of the gold carbene moiety via the formation of a tris-coordinated gold complex (i.e., D) [11].
![[1860-5397-9-227-1]](/bjoc/content/figures/1860-5397-9-227-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The impact of ligands on the ratio of 5a-OAc and 5a-H in the gold-catalyzed oxidation of 4a (reaction conditions: 5 mol % gold catalyst, 1.5 equiv of 3, DCE, rt, 3 h).
Figure 1: The impact of ligands on the ratio of 5a-OAc and 5a-H in the gold-catalyzed oxidation of 4a (reacti...
The scope of this alkyne oxidation/acetoxy migration reaction is outlined in Table 1. Acetates derived from primary/secondary propargylic alcohols with various substitution patterns and containing different functional groups were all allowed, although the tertiary counterpart underwent gold-catalyzed 3,3-rearrangement preferentially [21] and hence was not a viable substrate. Except entry 7, the gold-catalyzed oxidations proceeded with excellent regioselectivities (>25:1), and the desired α-acyloxy α,β-unsaturated ketones/aldehyde were isolated with fair to excellent yields. While the bulky catalyst Me4t-BuXPhosAuNTf2 [38] was used in entry 1 to obtain a better oxidation regioselectivity (28/1), it did not lead to a good ratio in the case of 4h (entry 7), where the oxidation regioisomer of type 5a’ was formed in 23% yield. This outcome is rationalized in the next paragraph. In the case of pivalate 3c with a terminal alkyne (entry 2), the use of this bulky acyl group instead of acetyl is to curtail the hydrolytic formation of the corresponding α-ketoaldehyde. In many cases the ratios of 5-OAc and 5-H were high with IPrAuNTf2 as the catalyst; for the ones with low selectivities, L1AuNTf2 offered again dramatic improvements (entries 5, 8 and 9) although at the expense of the geometric selectivities of the major product.
Table 1: Reaction scope studies for the formation of α-acetoxyenones from propargyl acetates.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-227-i5.svg?max-width=637&scale=1.0)
|
|||||||
| entry | 4 | 5 |
yieldb
ratioc time |
entry | 4 | 5 |
yieldb
ratioc |
|---|---|---|---|---|---|---|---|
| 1d |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-227-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-227-i7.svg?max-width=637&scale=1.0)
|
80%
14/1 3 h |
7d |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-227-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-227-i9.svg?max-width=637&scale=1.0)
|
62%
21/1h 5 h |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-227-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-227-i11.svg?max-width=637&scale=1.0)
|
74%e
>100:1 2.5 h |
8f |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-227-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-227-i13.svg?max-width=637&scale=1.0)
|
60%
>200/1 6.5 h |
| 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-227-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-227-i15.svg?max-width=637&scale=1.0)
|
86%
>50/1 12 h |
9f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-227-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-227-i17.svg?max-width=637&scale=1.0)
|
75%
>100/1 2.5 h |
| 4 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-227-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-227-i19.svg?max-width=637&scale=1.0)
|
85%
>50/1 9 h |
10 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-227-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-227-i21.svg?max-width=637&scale=1.0)
|
90%
>20/1 10 h |
| 5f,g |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-227-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-227-i23.svg?max-width=637&scale=1.0)
|
75%
>200/1 2.5 h |
11 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-227-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-227-i25.svg?max-width=637&scale=1.0)
|
84%
>35/1 5 h |
| 6 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-227-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-227-i27.svg?max-width=637&scale=1.0)
|
76%
33/1 7 h |
12 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-227-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-227-i29.svg?max-width=637&scale=1.0)
|
85%
>20/1 10 h |
a[4] = 0.05 M. bIsolated yield of 5-OAc. c5-OAc/5-H. dMe4t-BuXPhosAuNTf2 was used as the catalyst. eIPrAuCl/AgSbF6 as the catalyst, 3,5-dicholoropyridine N-oxide (2 equiv) as the oxidant, and DCM as the solvent. fL1AuCl/AgNTf2 used as catalyst. g10 mol % catalyst. hThe oxidation regioisomer of type 5a’ was formed in a 23% yield.
The excellent regioselectivities of gold-catalyzed oxidations of propargylic carboxylates, albeit unexpected, could be readily rationalized by invoking inductive polarization of the C–C triple bond by the electronegative carboxy group. Such polarization could be revealed by calculated natural charges via natural population analysis [39] and corroborated by experimentally detectable properties such as pKa [40] and 13C NMR [41]. We calculated the natural charges at the sp-hybridized carbons in 4a using the Density Functional Theory (B3LYP/6-31G*, Spartan06). The NC is 0.230 for the C(sp) distal to the carboxy group and −0.081 for the proximal C(sp), revealing a strong inductive effect that leads to a more electron-defficient distal alkyne end (Figure 2). This revelation is consistent with the 13C NMR chemical shifts of the alkynyl carbons. The observed regioselectivity can be ascribed to a selective attack of the nucleophilic oxidant to the more electrophilic distal C(sp). Notably, a recently published Pt-catalyzed hydrosilylation on a similar substrate showed a 3.7:1 regioselectivity [42]. This unexpectedly high selectivity with gold catalysis is attributed to the augmentation of the electronic bias of the C–C triple bond via the gold activation. The decreased regioselectivity with 4h (entry 7) is due to the counter polarization of the C–C triple bond by the propargylic BnO group. The better result with 4i also containing a similarly positioned BnO group (entry 8) is attributed to the synergistic effect of the steric bias [2].
![[1860-5397-9-227-2]](/bjoc/content/figures/1860-5397-9-227-2.svg?scale=1.8&max-width=1024&background=FFFFFF)
Figure 2: Natural charges at and the 13C chemical shifts of the alkynyl carbons in 4a.
Figure 2: Natural charges at and the 13C chemical shifts of the alkynyl carbons in 4a.
While a previous Pd catalysis [43] could also accomplish this transformation, the demonstrated scope is much limited, and the catalyst loading is 20%. With this oxidative gold catalysis, the propargyl esters, except those derived from tertiary alcohols, can be reliably converted into α-acyloxy α,β-unsaturated ketones/aldehydes.
Conclusion
We have realized a gold-catalyzed, highly regioselective oxidation of carboxylates of primary and secondary propargylic alcohols by utilizing inductive polarization of the C-C triple bond by the electron-withdrawing carboxy moiety. The α-oxo gold carbene intermediates generated can selectively undergo 1,2-acyloxy migrations over 1,2-C–H insertion. This inherent selectivity can be much enhanced by the use of our previously developed P,S-bidentate ligand, which enables the generation of tri-coordinated and less electrophilic gold carbene species. α-Acyloxy α,β-unsaturated ketones/aldehydes can be obtained with fair to excellent yields.
Supporting Information
| Supporting Information File 1: Experimental procedure, compound characterization, and NMR spectra. | ||
| Format: PDF | Size: 14.3 MB | Download |
References
-
Ye, L.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 3258–3259. doi:10.1021/ja100041e
Return to citation in text: [1] [2] -
Lu, B.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 14070–14072. doi:10.1021/ja1072614
Return to citation in text: [1] [2] [3] [4] -
Doyle, M. P.; McKevey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998.
Return to citation in text: [1] -
Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2903. doi:10.1021/cr0200217
Return to citation in text: [1] -
Taber, D. F. In Carbon-Carbon Σ-Bond Formation; Pattenden, G., Ed.; Pergamon Press: Oxford, UK, 1991; pp 1045–1062.
Return to citation in text: [1] -
Ye, L.; He, W.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 8550–8551. doi:10.1021/ja1033952
Return to citation in text: [1] -
He, W.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2011, 8482–8485. doi:10.1021/ja2029188
Return to citation in text: [1] -
Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624
Return to citation in text: [1] -
Wang, Y.; Ji, K.; Lan, S.; Zhang, L. Angew. Chem., Int. Ed. 2012, 51, 1915–1918. doi:10.1002/anie.201107561
Return to citation in text: [1] -
Ji, K.; Zhao, Y.; Zhang, L. Angew. Chem., Int. Ed. 2013, 52, 6508–6512. doi:10.1002/anie.201301601
Return to citation in text: [1] -
Luo, Y.; Ji, K.; Li, Y.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 17412–17415. doi:10.1021/ja307948m
Return to citation in text: [1] [2] [3] -
He, W.; Xie, L.; Xu, Y.; Xiang, J.; Zhang, L. Org. Biomol. Chem. 2012, 10, 3168–3171. doi:10.1039/c2ob25235j
Return to citation in text: [1] -
Bhunia, S.; Ghorpade, S.; Huple, D. B.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 2939–2942. doi:10.1002/anie.201108027
Return to citation in text: [1] -
Vasu, D.; Hung, H.-H.; Bhunia, S.; Gawade, S. A.; Das, A.; Liu, R.-S. Angew. Chem., Int. Ed. 2011, 50, 6911–6914. doi:10.1002/anie.201102581
Return to citation in text: [1] -
Henrion, G.; Chava, T. E. J.; Le Goff, X.; Gagosz, F. Angew. Chem., Int. Ed. 2013, 52, 6277–6882. doi:10.1002/anie.201301015
Return to citation in text: [1] -
Hashmi, A. S. K.; Wang, T.; Shi, S.; Rudolph, M. J. Org. Chem. 2012, 7761–7767. doi:10.1021/jo301381z
Return to citation in text: [1] -
Xu, M.; Ren, T.-T.; Li, C.-Y. Org. Lett. 2012, 14, 4902–4905. doi:10.1021/ol302238t
Return to citation in text: [1] -
Dateer, R. B.; Pati, K.; Liu, R.-S. Chem. Commun. 2012, 48, 7200–7202. doi:10.1039/c2cc33030j
Return to citation in text: [1] -
Qian, D.; Zhang, J. Chem. Commun. 2011, 47, 11152–11154. doi:10.1039/c1cc14788a
Return to citation in text: [1] -
Davies, P. W.; Cremonesi, A.; Martin, N. Chem. Commun. 2011, 47, 379–381. doi:10.1039/c0cc02736g
Return to citation in text: [1] -
Wang, S.; Zhang, G.; Zhang, L. Synlett 2010, 692–706. doi:10.1055/s-0029-1219527
Return to citation in text: [1] [2] -
Mamane, V.; Gress, T.; Krause, H.; Fuerstner, A. J. Am. Chem. Soc. 2004, 126, 8654–8655. doi:10.1021/ja048094q
Return to citation in text: [1] -
Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656–8657. doi:10.1021/ja0474695
Return to citation in text: [1] -
Murai, M.; Kitabata, S.; Okamoto, K.; Ohe, K. Chem. Commun. 2012, 48, 7622–7624. doi:10.1039/c2cc32628k
Return to citation in text: [1] -
Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 18002–18003. doi:10.1021/ja0552500
Return to citation in text: [1] -
Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804–16805. doi:10.1021/ja056419c
Return to citation in text: [1] -
Wang, S.; Zhang, L. J. Am. Chem. Soc. 2006, 128, 8414–8415. doi:10.1021/ja062777j
Return to citation in text: [1] -
Wang, S.; Zhang, L. M. Org. Lett. 2006, 8, 4585–4587. doi:10.1021/ol0618151
Return to citation in text: [1] -
Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; de Fremont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571
Return to citation in text: [1] -
Yu, M.; Li, G.; Wang, S.; Zhang, L. Adv. Synth. Catal. 2007, 349, 871–875. doi:10.1002/adsc.200600579
Return to citation in text: [1] -
Li, G.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2008, 130, 3740–3741. doi:10.1021/ja800001h
Return to citation in text: [1] -
Witham, C. A.; Mauleon, P.; Shapiro, N. D.; Sherry, B. D.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 5838–5839. doi:10.1021/ja071231+
Return to citation in text: [1] -
Cuenca, A. B.; Montserrai, S.; Hossain, K. M.; Mancha, G.; Lledós, A.; Medio-Simón, M.; Ujaque, G.; Asensio, G. Org. Lett. 2009, 11, 4906–4909. doi:10.1021/ol9020578
Return to citation in text: [1] -
Li, C.-W.; Pati, K.; Lin, G.-Y.; Sohel, S. M. A.; Hung, H.-H.; Liu, R.-S. Angew. Chem., Int. Ed. 2010, 49, 9891–9894. doi:10.1002/anie.201004647
Return to citation in text: [1] -
Lundgren, R. J.; Peters, B. D.; Alsabeh, P. G.; Stradiotto, M. Angew. Chem., Int. Ed. 2010, 49, 4071–4074. doi:10.1002/anie.201000526
Return to citation in text: [1] -
Hesp, K. D.; Stradiotto, M. J. Am. Chem. Soc. 2010, 132, 18026–18029. doi:10.1021/ja109192w
Return to citation in text: [1] -
Wang, Y.; Ji, K.; Lan, S.; Zhang, L. Angew. Chem., Int. Ed. 2012, 51, 1915–1918. doi:10.1002/anie.201107561
Return to citation in text: [1] -
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735–746. doi:10.1063/1.449486
Return to citation in text: [1] -
Gross, K. C.; Seybold, P. G.; Hadad, C. M. Int. J. Quant. Chem. 2002, 90, 445–458. doi:10.1002/qua.10108
Return to citation in text: [1] -
Levy, J. B. Struct. Chem. 1999, 10, 121–127. doi:10.1023/A:1022033330273
Return to citation in text: [1] -
Rooke, D. A.; Ferreira, E. M. Angew. Chem., Int. Ed. 2012, 51, 3225–3230. doi:10.1002/anie.201108714
Return to citation in text: [1] -
Bartels, A.; Mahrwald, R.; Müller, K. Adv. Synth. Catal. 2004, 346, 483–485. doi:10.1002/adsc.200303200
Return to citation in text: [1]
| 1. | Ye, L.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 3258–3259. doi:10.1021/ja100041e |
| 2. | Lu, B.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 14070–14072. doi:10.1021/ja1072614 |
| 6. | Ye, L.; He, W.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 8550–8551. doi:10.1021/ja1033952 |
| 7. | He, W.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2011, 8482–8485. doi:10.1021/ja2029188 |
| 8. | Ye, L.; He, W.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 3236–3239. doi:10.1002/anie.201007624 |
| 9. | Wang, Y.; Ji, K.; Lan, S.; Zhang, L. Angew. Chem., Int. Ed. 2012, 51, 1915–1918. doi:10.1002/anie.201107561 |
| 10. | Ji, K.; Zhao, Y.; Zhang, L. Angew. Chem., Int. Ed. 2013, 52, 6508–6512. doi:10.1002/anie.201301601 |
| 11. | Luo, Y.; Ji, K.; Li, Y.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 17412–17415. doi:10.1021/ja307948m |
| 12. | He, W.; Xie, L.; Xu, Y.; Xiang, J.; Zhang, L. Org. Biomol. Chem. 2012, 10, 3168–3171. doi:10.1039/c2ob25235j |
| 33. | Witham, C. A.; Mauleon, P.; Shapiro, N. D.; Sherry, B. D.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 5838–5839. doi:10.1021/ja071231+ |
| 3. | Doyle, M. P.; McKevey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998. |
| 4. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2903. doi:10.1021/cr0200217 |
| 5. | Taber, D. F. In Carbon-Carbon Σ-Bond Formation; Pattenden, G., Ed.; Pergamon Press: Oxford, UK, 1991; pp 1045–1062. |
| 34. | Cuenca, A. B.; Montserrai, S.; Hossain, K. M.; Mancha, G.; Lledós, A.; Medio-Simón, M.; Ujaque, G.; Asensio, G. Org. Lett. 2009, 11, 4906–4909. doi:10.1021/ol9020578 |
| 35. | Li, C.-W.; Pati, K.; Lin, G.-Y.; Sohel, S. M. A.; Hung, H.-H.; Liu, R.-S. Angew. Chem., Int. Ed. 2010, 49, 9891–9894. doi:10.1002/anie.201004647 |
| 2. | Lu, B.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 14070–14072. doi:10.1021/ja1072614 |
| 31. | Yu, M.; Li, G.; Wang, S.; Zhang, L. Adv. Synth. Catal. 2007, 349, 871–875. doi:10.1002/adsc.200600579 |
| 1. | Ye, L.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 3258–3259. doi:10.1021/ja100041e |
| 32. | Li, G.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2008, 130, 3740–3741. doi:10.1021/ja800001h |
| 2. | Lu, B.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 14070–14072. doi:10.1021/ja1072614 |
| 25. | Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 18002–18003. doi:10.1021/ja0552500 |
| 22. | Mamane, V.; Gress, T.; Krause, H.; Fuerstner, A. J. Am. Chem. Soc. 2004, 126, 8654–8655. doi:10.1021/ja048094q |
| 23. | Harrak, Y.; Blaszykowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656–8657. doi:10.1021/ja0474695 |
| 26. | Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804–16805. doi:10.1021/ja056419c |
| 27. | Wang, S.; Zhang, L. J. Am. Chem. Soc. 2006, 128, 8414–8415. doi:10.1021/ja062777j |
| 28. | Wang, S.; Zhang, L. M. Org. Lett. 2006, 8, 4585–4587. doi:10.1021/ol0618151 |
| 29. | Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442–1443. doi:10.1021/ja057327q |
| 30. | Marion, N.; Díez-González, S.; de Fremont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571 |
| 21. | Wang, S.; Zhang, G.; Zhang, L. Synlett 2010, 692–706. doi:10.1055/s-0029-1219527 |
| 13. | Bhunia, S.; Ghorpade, S.; Huple, D. B.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 2939–2942. doi:10.1002/anie.201108027 |
| 14. | Vasu, D.; Hung, H.-H.; Bhunia, S.; Gawade, S. A.; Das, A.; Liu, R.-S. Angew. Chem., Int. Ed. 2011, 50, 6911–6914. doi:10.1002/anie.201102581 |
| 15. | Henrion, G.; Chava, T. E. J.; Le Goff, X.; Gagosz, F. Angew. Chem., Int. Ed. 2013, 52, 6277–6882. doi:10.1002/anie.201301015 |
| 16. | Hashmi, A. S. K.; Wang, T.; Shi, S.; Rudolph, M. J. Org. Chem. 2012, 7761–7767. doi:10.1021/jo301381z |
| 17. | Xu, M.; Ren, T.-T.; Li, C.-Y. Org. Lett. 2012, 14, 4902–4905. doi:10.1021/ol302238t |
| 18. | Dateer, R. B.; Pati, K.; Liu, R.-S. Chem. Commun. 2012, 48, 7200–7202. doi:10.1039/c2cc33030j |
| 19. | Qian, D.; Zhang, J. Chem. Commun. 2011, 47, 11152–11154. doi:10.1039/c1cc14788a |
| 20. | Davies, P. W.; Cremonesi, A.; Martin, N. Chem. Commun. 2011, 47, 379–381. doi:10.1039/c0cc02736g |
| 24. | Murai, M.; Kitabata, S.; Okamoto, K.; Ohe, K. Chem. Commun. 2012, 48, 7622–7624. doi:10.1039/c2cc32628k |
| 11. | Luo, Y.; Ji, K.; Li, Y.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 17412–17415. doi:10.1021/ja307948m |
| 11. | Luo, Y.; Ji, K.; Li, Y.; Zhang, L. J. Am. Chem. Soc. 2012, 134, 17412–17415. doi:10.1021/ja307948m |
| 36. | Lundgren, R. J.; Peters, B. D.; Alsabeh, P. G.; Stradiotto, M. Angew. Chem., Int. Ed. 2010, 49, 4071–4074. doi:10.1002/anie.201000526 |
| 37. | Hesp, K. D.; Stradiotto, M. J. Am. Chem. Soc. 2010, 132, 18026–18029. doi:10.1021/ja109192w |
| 2. | Lu, B.; Li, C.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 14070–14072. doi:10.1021/ja1072614 |
| 43. | Bartels, A.; Mahrwald, R.; Müller, K. Adv. Synth. Catal. 2004, 346, 483–485. doi:10.1002/adsc.200303200 |
| 42. | Rooke, D. A.; Ferreira, E. M. Angew. Chem., Int. Ed. 2012, 51, 3225–3230. doi:10.1002/anie.201108714 |
| 39. | Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735–746. doi:10.1063/1.449486 |
| 40. | Gross, K. C.; Seybold, P. G.; Hadad, C. M. Int. J. Quant. Chem. 2002, 90, 445–458. doi:10.1002/qua.10108 |
| 21. | Wang, S.; Zhang, G.; Zhang, L. Synlett 2010, 692–706. doi:10.1055/s-0029-1219527 |
| 38. | Wang, Y.; Ji, K.; Lan, S.; Zhang, L. Angew. Chem., Int. Ed. 2012, 51, 1915–1918. doi:10.1002/anie.201107561 |
© 2013 Ji et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)