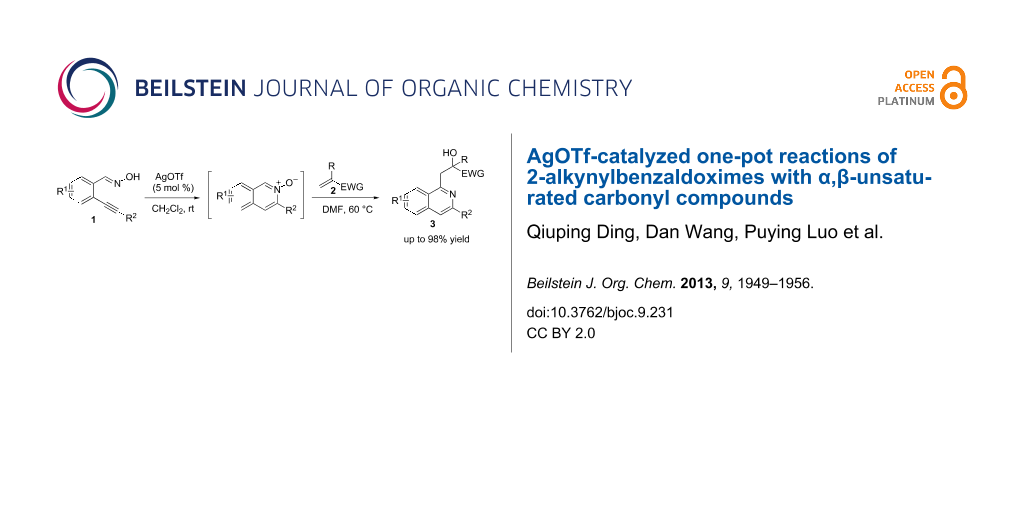
Abstract
AgOTf-catalyzed one-pot reactions of 2-alkynylbenzaldoximes with various α,β-unsaturated carbonyl compounds under mild conditions are described, which provides a facile and efficient pathway for the synthesis of 1-alkylated isoquinoline derivatives. The method tolerates a wide range of substrates and allows for the preparation of the products of interest in moderate to excellent yields.

Graphical Abstract
Introduction
One-pot combinations of multi-catalysis and multi-component cascade reactions [1-6], in which several bond-forming steps take place in a single operation, play an important role in atom-economical organic chemistry. A cascade reaction is the most efficient way for targeting fine chemicals, agrochemicals, pharmaceutical drugs, drug intermediates and ingredients by a one-pot reaction in environmentally and economically friendly synthetic processes. Isoquinoline derivatives, an important class of nitrogen-containing polycyclic heteroarenes, have attracted considerable attention because of their pharmacological activities, including antitumor, antifungal, antimalarial, antihypertensive and antihistaminic activity, and their photo- and electrochemical properties [7-15]. Over the past decade, there has been growing interest in the development of new methods for the construction of isoquinoline. For instance, Yamamoto [16-19], Larock [20-27], and Wu [28-35] have reported mild and efficient methodologies to synthesize substituted isoquinolines.
Despite the aforementioned versatile and efficient methods for the direct construction of isoquinolines, the selective functionalization of isoquinoline species is still a challenging task. Recently, there has been some progress in this aspect. Wu and co-workers described an efficient three-component reaction of a 2-alkynylbenzaldoxime and an α,β-unsaturated carbonyl compound with bromine or iodine monochloride under mild conditions, which generates the 1-alkylated isoquinolines in good to excellent yields [36]. Wu and co-workers also reported many other highly functionalized isoquinoline derivatives by cascade reactions in good yields under mild conditions, such as 1-aminoisoquinolines [37] and 1-(isoquinolin-1-yl)ureas [38,39]. Recently, Deng and co-workers also described a new Pd-catalyzed C–H oxidation system for the regioselective alkylation of isoquinoline N-oxide and its derivatives with sulfoxides for the synthesis of 1-alkylated isoquinolines [40].
We also reported the synthesis of 1-arylated 1,3-disubstituted isoquinoline N-oxides in a one-pot reaction characterized by a Ag-catalyzed intramolecular addition cyclization/Pd-catalyzed direct arylation of 2-alkynylbenzaldoximes [41]. Inspired by the key contributions from the groups of Wu [36-39] and Deng [40], we envisioned that 1-alkylated isoquinolines could be generated in a one-pot AgOTf-catalyzed cyclization/1,3-dipolar cycloaddition/rearrangement or fragmentation from 2-alkynylbenzaldoximes and α,β-unsaturated carbonyl compounds.
Based on previous results [36-39,41-43], we expect 2-alkynylbenzaldoxime 1 to easily convert at room temperature to isoquinoline N-oxide A by a AgOTf-catalyzed cyclization. Compound A produced in situ might undergo a 1,3-dipolar cycloaddition with α,β-unsaturated carbonyl compound 2 leading to 2,10b-dihydro-1H-isoxazolo[3,2-a]isoquinoline intermediate B [44,45], which may then suffer a rearrangement or fragmentation resulting in compound 3 (Scheme 1) [35,36,38,42]. To demonstrate the feasibility of this assumed route, we started to investigate the possibility of this one-pot process.
![[1860-5397-9-231-i1]](/bjoc/content/inline/1860-5397-9-231-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed route for the AgOTf-catalyzed one-pot reaction of 2-alkynylbenzaldoxime with an α,β-unsaturated carbonyl compound.
Scheme 1: Proposed route for the AgOTf-catalyzed one-pot reaction of 2-alkynylbenzaldoxime with an α,β-unsatu...
Results and Discussion
Initially, a set of experiments was carried out with 2-alkynylbenzaldoxime 1a and methyl methacrylate (2a) as model substrates in the presence of AgOTf (5 mol %). As expected, the reaction proceeded smoothly in CH2Cl2 at room temperature to afford the desired product 3a in 55% yield. We also tested other solvents, such as 1,4-dioxane, NMP, DMSO, DMA, toluene and DMF (Scheme 2). The solvent screening demonstrated that DMF was the best choice for the reaction at 60 °C. From these results, it was found that this one-pot process was highly efficient to construct 1-alkylated isoquinolines under very mild conditions.
![[1860-5397-9-231-i2]](/bjoc/content/inline/1860-5397-9-231-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1-alkylated isoquinoline 3a by a AgOTf-catalyzed one-pot reaction in different solvents.
Scheme 2: Synthesis of 1-alkylated isoquinoline 3a by a AgOTf-catalyzed one-pot reaction in different solvent...
With the optimized conditions in hand, the scope of the procedure was investigated for the reaction of 2-(p-tolylethynyl)benzaldehyde oxime (1b) with a series of α,β-unsaturated carbonyl compounds 2a–g (Table 1). In most cases, substrate 1b reacted with α,β-unsaturated carbonyl compounds 2 leading to the corresponding 1-alkylated isoquinolines in moderate to excellent yields. For instance, the reaction with 2b under standard conditions gave rise to the desired product 3c in 81% yield (Table 1, entry 2). An excellent yield was observed when butyl acrylate (2e) was utilized in the reaction (98% yield, Table 1, entry 5). When tert-butyl acrylate (2f) was employed, the reaction led to the formation of the desired 1-alkylated product 3g (80% yield, Table 1, entry 6) with a molar ratio of syn- and anti-isomers of ~1/6. It is noteworthy, that in other cases only a single product was observed. But-3-en-2-one (2g) was less reactive than the investigated acrylic acid esters and delivered the desired product only in moderate yield (40%, Table 1, entry 7). On the other hand, when substrate 1b was treated with acrylonitrile 2h under such conditions, the starting materials were recovered almost completely (Table 1, entry 8).
Table 1: AgOTf-catalyzed one-pot reactions of 2-(p-tolylethynyl)benzaldehyde oxime (1b) with α,β-unsaturated carbonyl compounds 2.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-231-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | 2 | Product 3 | Yielda (%) |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-231-i5.svg?max-width=637&scale=1.0)
2a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-231-i6.svg?max-width=637&scale=1.0)
3b |
70 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-231-i7.svg?max-width=637&scale=1.0)
2b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-231-i8.svg?max-width=637&scale=1.0)
3c |
81 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-231-i9.svg?max-width=637&scale=1.0)
2c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-231-i10.svg?max-width=637&scale=1.0)
3d |
50 |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-231-i11.svg?max-width=637&scale=1.0)
2d |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-231-i12.svg?max-width=637&scale=1.0)
3e |
61 |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-231-i13.svg?max-width=637&scale=1.0)
2e |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-231-i14.svg?max-width=637&scale=1.0)
3f |
98 |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-231-i15.svg?max-width=637&scale=1.0)
2f |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-231-i16.svg?max-width=637&scale=1.0)
3g |
80
(1/6)b |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-231-i17.svg?max-width=637&scale=1.0)
2g |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-231-i18.svg?max-width=637&scale=1.0)
3h |
40 |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-231-i19.svg?max-width=637&scale=1.0)
2h |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-231-i20.svg?max-width=637&scale=1.0)
3i |
– |
aIsolated yield based on 1b. bRatio of syn/anti, determined by 1H NMR.
Next, we examined the effect of substituents at the 2-alkynylbenzaldoxime 1. In most cases, 2-alkynylbenzaldoxime 1 reacted with acrylates 2 leading to the desired products 3 in moderate to good yields. For instance, reaction of 2-((4-methoxyphenyl)ethynyl)benzaldehyde oxime (1c) with methyl methacrylate (2a) under the conditions described above gave the desired product 3j in 75% yield (Table 2, entry 1). A better yield was obtained when substrate 1e was employed in the reaction (83% yield, Table 2, entry 3). The usage of 2-(cyclopropylethynyl)benzaldehyde oxime (1f) in the reaction led to a similar yield (80% yield, Table 2, entry 5). However, low yields were obtained when 2-(hex-1-yn-1-yl)benzaldehyde oxime (1g) reacted with methyl methacrylate (2a). In the case of substrate 1h (R2 = SiMe3) only desilyl product 3p was observed in poor yield due to the instability of the product. When R2 was changed to H (2-ethynylbenzaldehyde oxime (1i), Table 2, entry 8), there was no reaction at all. Good yields were obtained when 2-alkynylbenzaldoximes substituted with other electron-withdrawing groups (such as 1j and 1k) reacted with acrylate 2a (Table 2, entries 9–12). However, substrates with electron-donating groups attached on the aromatic ring of 2-alkynylbenzaldoxime (such as substrate 1l, Table 2, entry 13) did not afford a desired product.
Table 2: One-pot reactions of 2-alkynylbenzaldoximes 1 with acrylates 2.
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-231-i21.svg?max-width=637&scale=1.0)
|
||||
| Entry | 2-Alkynylbenzaldoxime 1 | 2 | Product 3 | Yielda (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-231-i22.svg?max-width=637&scale=1.0)
1c |
2a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-231-i23.svg?max-width=637&scale=1.0)
3j |
75 |
| 2 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-231-i24.svg?max-width=637&scale=1.0)
1d |
2a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-231-i25.svg?max-width=637&scale=1.0)
3k |
48 |
| 3 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-231-i26.svg?max-width=637&scale=1.0)
1e |
2a |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-231-i27.svg?max-width=637&scale=1.0)
3l |
83 |
| 4 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-231-i28.svg?max-width=637&scale=1.0)
1e |
2f |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-231-i29.svg?max-width=637&scale=1.0)
3m |
70
(1/4)b |
| 5 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-231-i30.svg?max-width=637&scale=1.0)
1f |
2a |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-231-i31.svg?max-width=637&scale=1.0)
3n |
80 |
| 6 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-231-i32.svg?max-width=637&scale=1.0)
1g |
2a |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-231-i33.svg?max-width=637&scale=1.0)
3o |
35 |
| 7 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-231-i34.svg?max-width=637&scale=1.0)
1h |
2a |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-231-i35.svg?max-width=637&scale=1.0)
3p |
12 |
| 8 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-231-i36.svg?max-width=637&scale=1.0)
1i |
2a |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-231-i37.svg?max-width=637&scale=1.0)
3p |
– |
| 9 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-231-i38.svg?max-width=637&scale=1.0)
1j |
2a |
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-231-i39.svg?max-width=637&scale=1.0)
3q |
80 |
| 10 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-9-231-i40.svg?max-width=637&scale=1.0)
1j |
2e |
![[Graphic 38]](/bjoc/content/inline/1860-5397-9-231-i41.svg?max-width=637&scale=1.0)
3r |
72 |
| 11 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-9-231-i42.svg?max-width=637&scale=1.0)
1j |
2f |
![[Graphic 40]](/bjoc/content/inline/1860-5397-9-231-i43.svg?max-width=637&scale=1.0)
3s |
71 |
| 12 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-9-231-i44.svg?max-width=637&scale=1.0)
1k |
2a |
![[Graphic 42]](/bjoc/content/inline/1860-5397-9-231-i45.svg?max-width=637&scale=1.0)
3t |
85 |
| 13 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-9-231-i46.svg?max-width=637&scale=1.0)
1l |
2a |
![[Graphic 44]](/bjoc/content/inline/1860-5397-9-231-i47.svg?max-width=637&scale=1.0)
3u |
– |
aIsolated yield based on 1b. bRatio of syn/anti, determined by 1H NMR.
Recently, 1-alkenylated isoquinoline 4 was synthesized via a Pd-mediated C–H bond activation approach in a one-pot reaction. The intermediate isoquinoline N-oxide A was produced in situ from 2-alkynylbenzaldoximes and reacted with the α,β-unsaturated carbonyl compound 2e to yield 1-alkenylated isoquinoline 4 (Scheme 3). This observation indicated that the Palladium-catalyzed alkenylation reaction mechanism might be similar to that described by Cui and Wu [46].
![[1860-5397-9-231-i3]](/bjoc/content/inline/1860-5397-9-231-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Pd-catalyzed one-pot alkenylation reaction of 2-alkynylbenzaldoxime 1a and butyl acrylate (2e).
Scheme 3: Pd-catalyzed one-pot alkenylation reaction of 2-alkynylbenzaldoxime 1a and butyl acrylate (2e).
Conclusion
In summary, we have demonstrated that one-pot reactions of 2-alkynylbenzaldoximes with α,β-unsaturated carbonyl compounds catalyzed by AgOTf occur smoothly under mild conditions. The present method provides a facile and efficient pathway for the synthesis of 1-alkylated isoquinoline derivatives in moderate to excellent yields with a wide range of substrates. The present one-pot catalyst system was also found to be applicable to the synthesis of 1-alkenylated isoquinoline derivatives.
Experimental
General
All reactions were performed in test tubes under a nitrogen atmosphere. Flash column chromatography was performed with silica gel (200–300 mesh). Analytical thin-layer chromatography was performed on glass plates pre-coated with 0.25 mm 230–400 mesh silica gel and impregnated with a fluorescent indicator (254 nm). Spots on thin-layer chromatography plates were visualized by exposure to ultraviolet light. Organic solutions were concentrated on rotary evaporators at 25–35 °C. Commercial reagents and solvents were used as received. 1H and 13C NMR spectra were recorded on a Bruker AV 400 spectrometer at 400 MHz (1H) and 100 MHz (13C) at ambient temperature. Chemical shifts are reported in parts per million (ppm) on the delta scale (δ) and referenced to tetramethylsilane (0 ppm). HRMS analyses were performed in ESI mode on a Bruker mass spectrometer.
General procedure for the AgOTf-catalyzed one-pot reactions of 2-alkynylbenzaldoximes 1 with α,β-unsaturated carbonyl compounds 2: A mixture of 2-alkynylbenzaldoximes 1 (0.3 mmol) and AgOTf (0.015 mmol, 5 mol %) in CH2Cl2 (2 mL) was stirred at room temperature for 2 h, until 2-alkynylbenzaldoxime 1 was completely consumed. The solvent was removed under reduced pressure. Then, α,β-unsaturated carbonyl compound 2 (1.5 mmol, 5.0 equiv) in DMF (1 mL) was added to the residue, and allowed to stir at 60 °C overnight under a nitrogen atmosphere. After completion of the reaction as indicated by TLC, the reaction was quenched by water and extracted with ethyl acetate. The organic layers were dried with anhydrous MgSO4, the solvent was evaporated under reduced pressure, and the residue was purified by column chromatography with EtOAc/petroleum ether (1:5, v/v) as an eluent to yield the desired products 3.
Procedure for the synthesis of 1-alkenylated isoquinoline 4 by a Pd-mediated C–H bond activation approach: A solution of 2-alkynylbenzaldoxime 1a (0.3 mmol) and AgOTf (0.015 mmol, 5 mol %) in CH2Cl2 (2 mL) was stirred at rt for 2 h. Then, the solvent was removed under reduced pressure. Subsequently, a solution of α,β-unsaturated carbonyl compound 2e (1.5 mmol, 5.0 equiv) and PdCl2(PhCN)2 (5 mol %) in NMP (1 mL) was added to the residue, and allowed to stir overnight at 110 °C under a nitrogen atmosphere. After completion of the reaction as indicated by TLC, the reaction was quenched by water and extracted with ethyl acetate. The organic layers were dried with anhydrous MgSO4, the solvent was evaporated under reduced pressure. The residue was purified by column chromatography with EtOAc/petroleum ether (1:3, v/v) as an eluent to yield the desired products 4. For details, see Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 4.2 MB | Download |
Acknowledgements
Financial supported from the National Natural Science Foundation of China (21002042), the Jiangxi Educational Committee (GJJ12169), the Project of Jiangxi Youth Scientist (20122BCB23012), and the Open Project Program of Key Laboratory of Functional Small Organic Molecule, the Ministry of Education, and the Jiangxi Normal University (No. KLFS-KF-201204 and KLFS-KF-201217) is gratefully acknowledged.
References
-
Cane, D. E. Chem. Rev. 1990, 90, 1089–1103. doi:10.1021/cr00105a002
Return to citation in text: [1] -
Tietze, L. F. Chem. Rev. 1996, 96, 115–136. doi:10.1021/cr950027e
Return to citation in text: [1] -
Tietze, L. F.; Modi, A. Med. Res. Rev. 2000, 20, 304–322. doi:10.1002/1098-1128(200007)20:4<304::AID-MED3>3.0.CO;2-8
Return to citation in text: [1] -
Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. doi:10.1002/anie.200601872
Return to citation in text: [1] -
Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem., Int. Ed. 2007, 46, 1570–1581. doi:10.1002/anie.200603129
Return to citation in text: [1] -
D’Souza, D. M.; Mueller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095–1108. doi:10.1039/b608235c
Return to citation in text: [1] -
Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds. The Chemistry and Biology of Isoquinoline Alkaloids; Springer Verlag: Berlin, Germany, 1985. doi:10.1007/978-3-642-70128-3
Return to citation in text: [1] -
Kartsev, V. G. Med. Chem. Res. 2004, 13, 325–336. doi:10.1007/s00044-004-0038-2
Return to citation in text: [1] -
Menachery, M. D.; Lavanier, G. L.; Wetherly, M. L.; Guinaudeau, H.; Shamma, M. J. Nat. Prod. 1986, 49, 745–778. doi:10.1021/np50047a001
Return to citation in text: [1] -
Baker, B. J. Alkaloids: Chem. Biol. Perspect. 1996, 10, 357–407. doi:10.1016/S0735-8210(96)80028-8
Return to citation in text: [1] -
Lundstroem, J. Alkaloids 1983, 21, 255–327.
Return to citation in text: [1] -
Croisy-Delcey, M.; Croisy, A.; Carrez, D.; Huel, C.; Chiaroni, A.; Ducrot, P.; Bisagni, E.; Jin, L.; Leclercq, G. Bioorg. Med. Chem. 2000, 8, 2629–2641. doi:10.1016/S0968-0896(00)00194-2
Return to citation in text: [1] -
Parenty, A. D. C.; Song, Y.-F.; Richmond, C. J.; Cronin, L. Org. Lett. 2007, 9, 2253–2256. doi:10.1021/ol070263z
Return to citation in text: [1] -
Abet, V.; Nuñez, A.; Mendicuti, F.; Burgos, C.; Alvarez-Builla, J. J. Org. Chem. 2008, 73, 8800–8807. doi:10.1021/jo801549u
Return to citation in text: [1] -
Ahmed, E.; Briseno, A. L.; Xia, Y.; Jenekhe, S. A. J. Am. Chem. Soc. 2008, 130, 1118–1119. doi:10.1021/ja077444g
Return to citation in text: [1] -
Fischer, D.; Tomeba, H.; Pahadi, N. K.; Patil, N. T.; Huo, Z.; Yamamoto, Y. J. Am. Chem. Soc. 2008, 130, 15720–15725. doi:10.1021/ja805326f
Return to citation in text: [1] -
Asao, N.; Yudha, S.; Nogami, S. T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2005, 44, 5526–5528. doi:10.1002/anie.200500795
Return to citation in text: [1] -
Asao, N.; Chan, C. S.; Takahashi, K.; Yamamoto, Y. Tetrahedron 2005, 61, 11322–11326. doi:10.1016/j.tet.2005.09.012
Return to citation in text: [1] -
Ohtaka, M.; Nakamura, H.; Yamamoto, Y. Tetrahedron Lett. 2004, 45, 7339–7341. doi:10.1016/j.tetlet.2004.08.008
Return to citation in text: [1] -
Huang, Q.; Larock, R. C. J. Org. Chem. 2003, 68, 980–988. doi:10.1021/jo0261303
Return to citation in text: [1] -
Dai, G.; Larock, R. C. J. Org. Chem. 2003, 68, 920–928. doi:10.1021/jo026294j
Return to citation in text: [1] -
Dai, G.; Larock, R. C. J. Org. Chem. 2002, 67, 7042–7047. doi:10.1021/jo026016k
Return to citation in text: [1] -
Huang, Q.; Hunter, J. A.; Larock, R. C. J. Org. Chem. 2002, 67, 3437–3444. doi:10.1021/jo020020e
Return to citation in text: [1] -
Roesch, K. R.; Larock, R. C. J. Org. Chem. 2002, 67, 86–94. doi:10.1021/jo010579z
Return to citation in text: [1] -
Roesch, K. R.; Zhang, H.; Larock, R. C. J. Org. Chem. 2001, 66, 8042–8051. doi:10.1021/jo0105540
Return to citation in text: [1] -
Roesch, K. R.; Larock, R. C. Org. Lett. 1999, 1, 553–556. doi:10.1021/ol990067v
Return to citation in text: [1] -
Dai, G.; Larock, R. C. Org. Lett. 2001, 3, 4035–4038. doi:10.1021/ol0102085
Return to citation in text: [1] -
Chen, Z.; Ding, Q.; Yu, X.; Wu, J. Adv. Synth. Catal. 2009, 351, 1692–1698. doi:10.1002/adsc.200900131
Return to citation in text: [1] -
Chen, Z.; Su, M.; Yu, X.; Wu, J. Org. Biomol. Chem. 2009, 7, 4641–4646. doi:10.1039/b914265g
Return to citation in text: [1] -
Chen, Z.; Yang, X.; Wu, J. Chem. Commun. 2009, 45, 3469–3471. doi:10.1039/b904498a
Return to citation in text: [1] -
Yu, X.; Chen, Z.; Yang, X.; Wu, J. J. Comb. Chem. 2010, 12, 374–378. doi:10.1021/cc1000314
Return to citation in text: [1] -
Yu, X.; Wu, J. J. Comb. Chem. 2009, 11, 895–899. doi:10.1021/cc900079s
Return to citation in text: [1] -
Yu, X.; Yang, X.; Wu, J. Org. Biomol. Chem. 2009, 7, 4526–4530. doi:10.1039/b913409c
Return to citation in text: [1] -
Yu, X.; Ding, Q.; Chen, Z.; Wu, J. Tetrahedron Lett. 2009, 50, 4279–4282. doi:10.1016/j.tetlet.2009.05.013
Return to citation in text: [1] -
Ding, Q.; Wang, Z.; Wu, J. J. Org. Chem. 2009, 74, 921–924. doi:10.1021/jo802076k
Return to citation in text: [1] [2] -
Ye, S.; Gao, K.; Wu, J. Adv. Synth. Catal. 2010, 352, 1746–1751. doi:10.1002/adsc.201000080
Return to citation in text: [1] [2] [3] [4] -
Zheng, D.; Chen, Z.; Liu, J.; Wu, J. Org. Biomol. Chem. 2011, 9, 4763–4765. doi:10.1039/c1ob05582h
Return to citation in text: [1] [2] [3] -
Ye, S.; Wang, H.; Wu, J. Eur. J. Org. Chem. 2010, 6436–6439. doi:10.1002/ejoc.201001040
Return to citation in text: [1] [2] [3] [4] -
Ye, S.; Wang, H.; Wu, J. ACS Comb. Sci. 2011, 13, 120–125. doi:10.1021/co100026y
Return to citation in text: [1] [2] [3] -
Yao, B.; Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H.; Wang, M.-K.; Tang, R.-Y.; Zhang, X.-G.; Deng, C.-L. Adv. Synth. Catal. 2012, 354, 1890–1896. doi:10.1002/adsc.201101009
Return to citation in text: [1] [2] -
Ding, Q.; Wang, D.; Sang, X.; Lin, Y.; Peng, Y. Tetrahedron 2012, 68, 8869–8874. doi:10.1016/j.tet.2012.08.039
Return to citation in text: [1] [2] -
Chen, Z.; Yu, X.; Su, M.; Yang, X.; Wu, J. Adv. Synth. Catal. 2009, 351, 2702–2708. doi:10.1002/adsc.200900442
Return to citation in text: [1] [2] -
Yeom, H.-S.; Kim, S.; Shin, S. Synlett 2008, 924–928. doi:10.1055/s-2008-1042936
Return to citation in text: [1] -
Gao, Z.-X.; Wang, M.; Wang, S.; Yao, Z.-J. Org. Lett. 2009, 11, 3678–3681. doi:10.1021/ol901511x
Return to citation in text: [1] -
Huisgen, R.; Seidl, H.; Wulff, J. Chem. Ber. 1969, 102, 915–925. doi:10.1002/cber.19691020325
Return to citation in text: [1] -
Wu, J.; Cui, X.; Chen, L.; Jiang, G.; Wu, Y. J. Am. Chem. Soc. 2009, 131, 13888–13889. doi:10.1021/ja902762a
Return to citation in text: [1]
| 1. | Cane, D. E. Chem. Rev. 1990, 90, 1089–1103. doi:10.1021/cr00105a002 |
| 2. | Tietze, L. F. Chem. Rev. 1996, 96, 115–136. doi:10.1021/cr950027e |
| 3. | Tietze, L. F.; Modi, A. Med. Res. Rev. 2000, 20, 304–322. doi:10.1002/1098-1128(200007)20:4<304::AID-MED3>3.0.CO;2-8 |
| 4. | Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. doi:10.1002/anie.200601872 |
| 5. | Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem., Int. Ed. 2007, 46, 1570–1581. doi:10.1002/anie.200603129 |
| 6. | D’Souza, D. M.; Mueller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095–1108. doi:10.1039/b608235c |
| 28. | Chen, Z.; Ding, Q.; Yu, X.; Wu, J. Adv. Synth. Catal. 2009, 351, 1692–1698. doi:10.1002/adsc.200900131 |
| 29. | Chen, Z.; Su, M.; Yu, X.; Wu, J. Org. Biomol. Chem. 2009, 7, 4641–4646. doi:10.1039/b914265g |
| 30. | Chen, Z.; Yang, X.; Wu, J. Chem. Commun. 2009, 45, 3469–3471. doi:10.1039/b904498a |
| 31. | Yu, X.; Chen, Z.; Yang, X.; Wu, J. J. Comb. Chem. 2010, 12, 374–378. doi:10.1021/cc1000314 |
| 32. | Yu, X.; Wu, J. J. Comb. Chem. 2009, 11, 895–899. doi:10.1021/cc900079s |
| 33. | Yu, X.; Yang, X.; Wu, J. Org. Biomol. Chem. 2009, 7, 4526–4530. doi:10.1039/b913409c |
| 34. | Yu, X.; Ding, Q.; Chen, Z.; Wu, J. Tetrahedron Lett. 2009, 50, 4279–4282. doi:10.1016/j.tetlet.2009.05.013 |
| 35. | Ding, Q.; Wang, Z.; Wu, J. J. Org. Chem. 2009, 74, 921–924. doi:10.1021/jo802076k |
| 35. | Ding, Q.; Wang, Z.; Wu, J. J. Org. Chem. 2009, 74, 921–924. doi:10.1021/jo802076k |
| 36. | Ye, S.; Gao, K.; Wu, J. Adv. Synth. Catal. 2010, 352, 1746–1751. doi:10.1002/adsc.201000080 |
| 38. | Ye, S.; Wang, H.; Wu, J. Eur. J. Org. Chem. 2010, 6436–6439. doi:10.1002/ejoc.201001040 |
| 42. | Chen, Z.; Yu, X.; Su, M.; Yang, X.; Wu, J. Adv. Synth. Catal. 2009, 351, 2702–2708. doi:10.1002/adsc.200900442 |
| 20. | Huang, Q.; Larock, R. C. J. Org. Chem. 2003, 68, 980–988. doi:10.1021/jo0261303 |
| 21. | Dai, G.; Larock, R. C. J. Org. Chem. 2003, 68, 920–928. doi:10.1021/jo026294j |
| 22. | Dai, G.; Larock, R. C. J. Org. Chem. 2002, 67, 7042–7047. doi:10.1021/jo026016k |
| 23. | Huang, Q.; Hunter, J. A.; Larock, R. C. J. Org. Chem. 2002, 67, 3437–3444. doi:10.1021/jo020020e |
| 24. | Roesch, K. R.; Larock, R. C. J. Org. Chem. 2002, 67, 86–94. doi:10.1021/jo010579z |
| 25. | Roesch, K. R.; Zhang, H.; Larock, R. C. J. Org. Chem. 2001, 66, 8042–8051. doi:10.1021/jo0105540 |
| 26. | Roesch, K. R.; Larock, R. C. Org. Lett. 1999, 1, 553–556. doi:10.1021/ol990067v |
| 27. | Dai, G.; Larock, R. C. Org. Lett. 2001, 3, 4035–4038. doi:10.1021/ol0102085 |
| 46. | Wu, J.; Cui, X.; Chen, L.; Jiang, G.; Wu, Y. J. Am. Chem. Soc. 2009, 131, 13888–13889. doi:10.1021/ja902762a |
| 16. | Fischer, D.; Tomeba, H.; Pahadi, N. K.; Patil, N. T.; Huo, Z.; Yamamoto, Y. J. Am. Chem. Soc. 2008, 130, 15720–15725. doi:10.1021/ja805326f |
| 17. | Asao, N.; Yudha, S.; Nogami, S. T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2005, 44, 5526–5528. doi:10.1002/anie.200500795 |
| 18. | Asao, N.; Chan, C. S.; Takahashi, K.; Yamamoto, Y. Tetrahedron 2005, 61, 11322–11326. doi:10.1016/j.tet.2005.09.012 |
| 19. | Ohtaka, M.; Nakamura, H.; Yamamoto, Y. Tetrahedron Lett. 2004, 45, 7339–7341. doi:10.1016/j.tetlet.2004.08.008 |
| 36. | Ye, S.; Gao, K.; Wu, J. Adv. Synth. Catal. 2010, 352, 1746–1751. doi:10.1002/adsc.201000080 |
| 37. | Zheng, D.; Chen, Z.; Liu, J.; Wu, J. Org. Biomol. Chem. 2011, 9, 4763–4765. doi:10.1039/c1ob05582h |
| 38. | Ye, S.; Wang, H.; Wu, J. Eur. J. Org. Chem. 2010, 6436–6439. doi:10.1002/ejoc.201001040 |
| 39. | Ye, S.; Wang, H.; Wu, J. ACS Comb. Sci. 2011, 13, 120–125. doi:10.1021/co100026y |
| 41. | Ding, Q.; Wang, D.; Sang, X.; Lin, Y.; Peng, Y. Tetrahedron 2012, 68, 8869–8874. doi:10.1016/j.tet.2012.08.039 |
| 42. | Chen, Z.; Yu, X.; Su, M.; Yang, X.; Wu, J. Adv. Synth. Catal. 2009, 351, 2702–2708. doi:10.1002/adsc.200900442 |
| 43. | Yeom, H.-S.; Kim, S.; Shin, S. Synlett 2008, 924–928. doi:10.1055/s-2008-1042936 |
| 7. | Phillipson, J. D.; Roberts, M. F.; Zenk, M. H., Eds. The Chemistry and Biology of Isoquinoline Alkaloids; Springer Verlag: Berlin, Germany, 1985. doi:10.1007/978-3-642-70128-3 |
| 8. | Kartsev, V. G. Med. Chem. Res. 2004, 13, 325–336. doi:10.1007/s00044-004-0038-2 |
| 9. | Menachery, M. D.; Lavanier, G. L.; Wetherly, M. L.; Guinaudeau, H.; Shamma, M. J. Nat. Prod. 1986, 49, 745–778. doi:10.1021/np50047a001 |
| 10. | Baker, B. J. Alkaloids: Chem. Biol. Perspect. 1996, 10, 357–407. doi:10.1016/S0735-8210(96)80028-8 |
| 11. | Lundstroem, J. Alkaloids 1983, 21, 255–327. |
| 12. | Croisy-Delcey, M.; Croisy, A.; Carrez, D.; Huel, C.; Chiaroni, A.; Ducrot, P.; Bisagni, E.; Jin, L.; Leclercq, G. Bioorg. Med. Chem. 2000, 8, 2629–2641. doi:10.1016/S0968-0896(00)00194-2 |
| 13. | Parenty, A. D. C.; Song, Y.-F.; Richmond, C. J.; Cronin, L. Org. Lett. 2007, 9, 2253–2256. doi:10.1021/ol070263z |
| 14. | Abet, V.; Nuñez, A.; Mendicuti, F.; Burgos, C.; Alvarez-Builla, J. J. Org. Chem. 2008, 73, 8800–8807. doi:10.1021/jo801549u |
| 15. | Ahmed, E.; Briseno, A. L.; Xia, Y.; Jenekhe, S. A. J. Am. Chem. Soc. 2008, 130, 1118–1119. doi:10.1021/ja077444g |
| 44. | Gao, Z.-X.; Wang, M.; Wang, S.; Yao, Z.-J. Org. Lett. 2009, 11, 3678–3681. doi:10.1021/ol901511x |
| 45. | Huisgen, R.; Seidl, H.; Wulff, J. Chem. Ber. 1969, 102, 915–925. doi:10.1002/cber.19691020325 |
| 40. | Yao, B.; Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H.; Wang, M.-K.; Tang, R.-Y.; Zhang, X.-G.; Deng, C.-L. Adv. Synth. Catal. 2012, 354, 1890–1896. doi:10.1002/adsc.201101009 |
| 36. | Ye, S.; Gao, K.; Wu, J. Adv. Synth. Catal. 2010, 352, 1746–1751. doi:10.1002/adsc.201000080 |
| 37. | Zheng, D.; Chen, Z.; Liu, J.; Wu, J. Org. Biomol. Chem. 2011, 9, 4763–4765. doi:10.1039/c1ob05582h |
| 38. | Ye, S.; Wang, H.; Wu, J. Eur. J. Org. Chem. 2010, 6436–6439. doi:10.1002/ejoc.201001040 |
| 39. | Ye, S.; Wang, H.; Wu, J. ACS Comb. Sci. 2011, 13, 120–125. doi:10.1021/co100026y |
| 38. | Ye, S.; Wang, H.; Wu, J. Eur. J. Org. Chem. 2010, 6436–6439. doi:10.1002/ejoc.201001040 |
| 39. | Ye, S.; Wang, H.; Wu, J. ACS Comb. Sci. 2011, 13, 120–125. doi:10.1021/co100026y |
| 40. | Yao, B.; Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H.; Wang, M.-K.; Tang, R.-Y.; Zhang, X.-G.; Deng, C.-L. Adv. Synth. Catal. 2012, 354, 1890–1896. doi:10.1002/adsc.201101009 |
| 37. | Zheng, D.; Chen, Z.; Liu, J.; Wu, J. Org. Biomol. Chem. 2011, 9, 4763–4765. doi:10.1039/c1ob05582h |
| 36. | Ye, S.; Gao, K.; Wu, J. Adv. Synth. Catal. 2010, 352, 1746–1751. doi:10.1002/adsc.201000080 |
| 41. | Ding, Q.; Wang, D.; Sang, X.; Lin, Y.; Peng, Y. Tetrahedron 2012, 68, 8869–8874. doi:10.1016/j.tet.2012.08.039 |
© 2013 Ding et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)