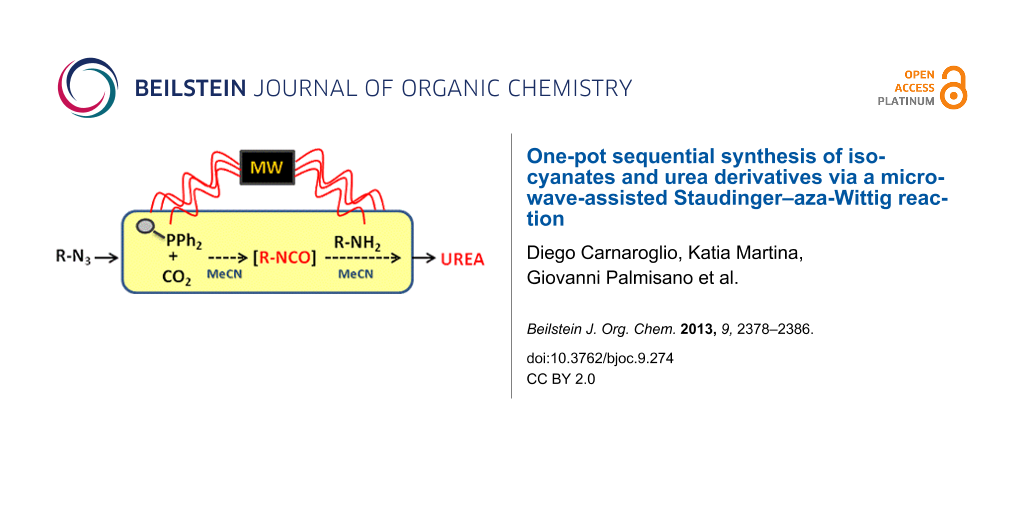
Abstract
A fast and efficient protocol for the synthesis of N,N'-disubstituted urea derivatives from alkyl halides and primary or secondary amines has been developed. The synthetic pathway combines nucleophilic substitutions and a Staudinger–aza-Wittig reaction in the presence of polymer-bound diphenylphosphine under 14 bar of CO2 pressure and has been performed in a one-pot two-step process. The protocol has been optimized under microwave irradiation and the scale-up experiment has been conducted under conventional conditions in a Parr reactor. The final compounds were isolated after simple filtration in almost quantitative overall yields which makes this procedure facile and rapid to execute.
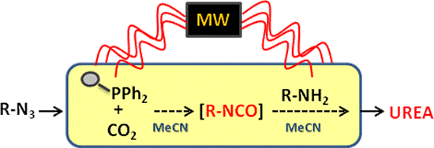
Graphical Abstract
Introduction
The industrial and commercial impact of isocyanates (R–NCO) is steadily growing. In particular, the polyurethane output has undergone yearly increases of 5% over the last decade [1]. Isocyanates play a relevant role as chemical intermediates in the manufacturing of thermoplastic foams, elastomers, adhesives, agrochemicals and pharmaceuticals. The isocyanate group is also widely used as a precursor to several bioactive compounds and drugs that contain urea and carbamate moieties [2]. Isocyanates were discovered by Wurtz in 1849 [3], and more than 20 methods for the preparation of R–NCO have now been listed and classified according to the reactions involved [4,5]. The old procedure that entails reactions between primary or secondary amines or amides with phosgene, is the most commonly used and it is described in papers [6,7] and reviews [8]. The main drawbacks of this method are the extremely high toxicity of phosgene and the generation of a large amount of corrosive HCl. Moreover, the high temperature required by this process (>250 °C) makes the synthesis of lower molecular weight compounds impossible.
Apart from the catalytic carbonylation of nitro compounds, which is one of the most interesting alternatives for the synthesis of aromatic isocyanates, other alternative greener, non-phosgene routes to isocyanates have also been developed [9]. Of these, the Curtius, Hoffman and Lossen rearrangements have been used quite often in the past and are still used for specific applications [10-13]. The Staudinger–aza-Wittig reactions have played a pivotal role in the construction of cyclic and acyclic compounds [14-19]. The replacement of phosgene by carbon dioxide (CO2), which is nontoxic, abundant, and economical, is the main advantage of this reaction. The mechanism of this transformation passes through iminophosphoranes that are versatile intermediates and can react with CO2 to generate isocyanates [20]. This reaction is compatible with a large number of functional groups and therefore has various uses in organic synthesis and can also be exploited for the preparation of heterocyclic compounds. Isocyanate derivatives can be generally obtained in good yields. However, it is necessary to avoid the traditional triphenylphosphine to obtain high purity products [21].
So called “enabling techniques”, mainly non-conventional energy sources such as microwaves (MW) and ultrasound (US), can dramatically enhance reaction rates in organic synthesis [22,23]. Kinetics and yields of any chemical modification are strongly improved by the optimal heat and mass transfer provided by dielectric heating and sonochemical conditions [24,25]. In general, MW in organic synthesis is a valid response to problems regarding long reaction times and a high reagent excess. The use of dielectric heating to promote chemical reactions has become well established as a reliable technique which can be applied on a range of scales [26]. Despite MW irradiation being commonly used in organic synthesis, only few publications describe this technique with gaseous reagents in closed vessels and in heterogeneous gas-phase reactions that are important for industrial processes [27-31]. The aim of the present work is the development of new green and efficient synthetic procedures for easier access to isocyanates and urea libraries using a renewable carbon resource like CO2. Since CO2 requires a large energy input to be transformed [32], we have studied a synthetic procedure which uses a MW reactor (SynthWave by Milestone) and is well suited for parallel syntheses at any reaction temperature and gas pressure (up to 300 °C and 200 bar). We aimed to successfully reduce the reaction time and the reagent excess, and to employ poorly reactive substrates and volatile, solid and supported reagents. In summary, we herein report an optimized protocol for a MW-assisted Staudinger–aza-Wittig reaction with polymer-bound diphenylphosphine (PS-PPh2) in a CO2 atmosphere. The study also aimed to prepare a series of symmetric and asymmetric alkyl/aryl urea derivatives in a one-pot, sequential synthesis of urea derivatives from alkyl bromides. With the aim to verify the feasibility of the method under conventional heating, the protocol was tested in a Parr reactor (90 mL) for an easier scale-up.
Results and Discussion
The Staudinger–aza-Wittig reaction is extremely versatile and can be used for the synthesis of many products. However, the byproduct of this reaction is triphenylphosphine oxide, which is difficult to remove. It is known that this reaction can be performed in a heterogeneous system using PS-PPh2. Despite the higher costs for the reagent, the use of PS-PPh2 has the advantage of a much easier reaction work-up [33-38]. A polymer regeneration procedure was described by Marsura et al. [35], however a quasi-stoichiometric amount makes the recycling step unneccessary. With the aim to overcome cost limitations we recently described the preparation of triphenylphosphine-loaded cross-liked cyclodextrin complexes as recyclable green catalyst [39]. The reactivity of polystyrene-supported reagents strongly depends on the choice of solvent that can influence polymer swelling [40]. Solvent choice is therefore an important issue as it must allow the supported reagent to work in a friendly environment and, at the same time, facilitate the reaction outcome. In the first part of this work, we have focused on the development of a MW promoted protocol for isocyanate synthesis with the aim of reducing reaction time and decreasing the amount of solid supported PS-PPh2, which is usually added in large excess. The conversion of benzyl azide to benzyl isocyanate (Scheme 1) was selected as the model reaction and it was performed both under conventional conditions and under MW irradiation. Various solvents were compared at a number of temperatures thanks to the versatility of the SynthWave reactor, which provides multiple-sample racks. Experiments were performed at 90, 70 and 50 °C at 14.5 bar of CO2 pressure.
![[1860-5397-9-274-i1]](/bjoc/content/inline/1860-5397-9-274-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of benzyl isocyanate.
Scheme 1: Synthesis of benzyl isocyanate.
As shown in Table 1, the reaction showed complete conversion when performed in toluene at 70 °C. The purity of the compound was also slightly higher than that of the reacition at 90 °C. The conversion was not complete at 50 °C. Conversion was low in THF and the isocyanate was present only as trace among side-products. MeCN, like toluene allowed a higher conversion and a higher yield to be achieved, even in comparison with DMF. In MeCN, there was a complete conversion even at 50 °C in 4 h. The influence of the CO2 pressure on the reaction rate was evident by the poor conversion (40%) that was observed when the reaction was performed with 1 bar CO2 at 50 °C in an oil bath. In contrast, the conversion reached 93% in a Parr reactor with 14 bar CO2. At room temperature with 1 bar CO2 the reaction occurs within 24 h.
Table 1: Synthesis of benzyl isocyanate.a
| entry | solvent | reaction conditions | conversionb(%) | yieldb(%) |
|---|---|---|---|---|
| 1 | toluene | 90 °C, MW, CO2 (14.5 bar) | >99 | 75 |
| 2 | toluene | 70 °C, MW, CO2 (14.5 bar) | 96 | 84 |
| 6 | toluene | 50 °C, MW, CO2 (14.5 bar) | 82 | 78 |
| 3 | THF | 70 °C, MW, CO2 (14.5 bar) | 50 | 25 |
| 4 | DMF | 70 °C, MW, CO2 (14.5 bar) | 95 | 80 |
| 5 | MeCN | 70 °C, MW, CO2 (14.5 bar) | >99 | 85 |
| 7 | MeCN | 50 °C, MW, CO2 (14.5 bar) | >99 | 94 |
| 8c | MeCN | 50 °C, CO2 (1 bar) | 41 | 25 |
| 9 | MeCN | rt, CO2 (1 bar) | 25 (95)d | 21 (85)d |
| 10e | MeCN | 50 °C, CO2 (14 bar) | 93 | 89 |
aUnless otherwise stated, reactions were performed in the presence of PS-PPh2 (5 equiv), reaction time 4 h. bDetermined by GC–MS. cThe reaction was performed in an oil bath. dReaction time 24 h. eThe reaction was performed in a Parr reactor.
In order to optimize the reaction conditions, another study was pursued in MeCN and toluene, and a number of different reactions were performed at 50 °C. As shown in Table 2, we confirm that the reaction was faster in MeCN than in toluene and that even full conversion was obtained after 1.5 h in many cases. An important goal was to reduce the PS-PPh2 excess from 5 to 1.5 equiv, with excellent results only in MeCN (Table 2) under MW irradiation. When the reaction was carried out in the Parr reactor, the highest conversion was 93% with 2 equiv PS-PPh2.
Table 2: Synthesis of benzyl isocyanate.a
| entry | solvent | PS-PPh2 (equiv) | time (h) | conversionb (%) | yieldb (%) |
|---|---|---|---|---|---|
| 1 | toluene | 5 | 4 | 82 | 78 |
| 2 | toluene | 5 | 2 | 71 | 71 |
| 3 | toluene | 2 | 2 | 66 | 54 |
| 4 | MeCN | 5 | 4 | >99 | 94 |
| 5 | MeCN | 5 | 2 | >99 | 96 |
| 6 | MeCN | 5 | 1.5 | >99 | 97 |
| 7 | MeCN | 5 | 1 | 80 | 76 |
| 8 | MeCN | 3 | 1.5 | >99 | 97 |
| 9 | MeCN | 2 | 1.5 | >99 | 95 |
| 10c | MeCN | 2 | 1.5 | 93 | 88 |
| 11 | MeCN | 1.5 | 1.5 | >99 | 98 |
| 12c | MeCN | 1.5 | 1.5 | 88 | 82 |
| 13 | MeCN | 1 | 1.5 | 80 | 76 |
aReactions were carried out in a MW reactor: 1, PS-PPh2, CO2 (14.5 bar) 50 °C. bDetermined by GC–MS. cReactions were carried out in a Parr reactor (90 mL): 1, PS-PPh2, CO2 (14 bar) 50 °C.
To confirm the versatility of our protocol, the method was extended to include a number of different substrates and the obtained isocyanates were also used for the synthesis of urea derivatives via the reaction with (±)-1-phenylethylamine 3 (see Scheme in Table 3). Seven different azido-derivatives were synthesized from the alkyl halide and reacted via Staudinger–aza-Wittig reactions. After the PS-PPh2 was filtered, the isocyanate solution was directly subjected to an addition of 2 equiv of 3 and heated up to 70 °C in the MW reactor for 3 h. Primary alkyl and benzyl azide derivatives were compared to secondary ones, and the compatibility of the protocol towards different functional groups was also considered. The results obtained are given in Table 3 and show the yield of urea derivatives after purification from the amine excess by using Dowex® 50WX8-200. This synthetic protocol is versatile and highly efficient with both primary and secondary azido derivatives as well as alkoxy and amido groups. The one-pot, two-step procedure afforded urea derivatives in high yield and purity via the isocyanate intermediate. To broaden the scope of the study, the reaction was repeated in a bigger scale (80 mL) in a Parr reactor at the same pressure. Despite the good conversion (about 93%), the product purity was slightly lower than that of the MW-assisted reaction.
Table 3: Synthesis optimization of urea derivatives.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-274-i2.svg?max-width=637&scale=1.0)
|
|||
| entry | R–N3 | product | yieldb (%) |
|---|---|---|---|
| 1 | 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-274-i3.svg?max-width=637&scale=1.0)
10 |
98 |
| 1c | 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-274-i4.svg?max-width=637&scale=1.0)
10 |
79 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-274-i5.svg?max-width=637&scale=1.0)
4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-274-i6.svg?max-width=637&scale=1.0)
11 |
90 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-274-i7.svg?max-width=637&scale=1.0)
5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-274-i8.svg?max-width=637&scale=1.0)
12 |
92 |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-274-i9.svg?max-width=637&scale=1.0)
6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-274-i10.svg?max-width=637&scale=1.0)
13 |
97 |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-274-i11.svg?max-width=637&scale=1.0)
7 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-274-i12.svg?max-width=637&scale=1.0)
14 |
97 |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-274-i13.svg?max-width=637&scale=1.0)
8 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-274-i14.svg?max-width=637&scale=1.0)
15 |
98 |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-274-i15.svg?max-width=637&scale=1.0)
9 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-274-i16.svg?max-width=637&scale=1.0)
16 |
94 |
aReactions were carried out in a MW reactor: azido derivative, PS-PPh2 (1.5 equiv), CO2 (14.5 bar), 50 °C, 1.5 h, then 3 (2 equiv) 70 °C, 3 h. bIsolated yield. cThe reaction was performed in a Parr reactor.
To expand the scope of this method, a sequential one-pot synthesis from the alkyl bromide to the urea derivative was carried out without isolating the intermediates. The aim of this part of the work was the synthesis of the azido derivatives and their subsequent conversion to urea via the Staudinger–aza-Wittig reaction and one-pot amine addition. The combination of reactants in a one-pot fashion can lead to undesired side-product formation and, consequently, a lower yield. Therefore, the choice of the right solvent and reaction conditions is the key to the success of this transformation.
Our initial attempts focused on the synthesis of azido derivatives in MeCN. Although generally performed in DMF, the SN2 reaction of alkyl bromide with NaN3 can also be performed in MeCN. NaN3 is insoluble in MeCN at room temperature (<0.005 g/100 mL), but its solubility increases at higher temperatures. Furthermore, NaBr generated during the nucleophilic substitution is insoluble and can be removed by filtration as can the NaN3 excess. These factors allow the work-up procedure to be simplified and pure azido derivative solutions were obtained by filtration. Benzyl azide was successfully obtained from benzyl bromide after the reaction in a MW reactor at 95 °C for 3 h. After filtration, the benzyl azide solution was directly converted into the urea compound by MW irradiation at 70 °C for 3 h in the presence of CO2, PS-PPh2 and benzylamine. The desired product was obtained in almost quantitative yields.
The robustness of this MW-promoted sequential one-pot procedure was established by synthesizing a set of 13 different urea derivatives. A small set of five different primary and secondary alkyl and benzyl azides were synthesized. Besides the azido derivatives synthesized from 17–19, the two volatile azides synthesized from 20 and 21 (n-butyl and allyl azide, respectively) were obtained under N2 pressure. After filtration they were converted into the urea compound. The procedure was performed in parallel and under 14.5 bar of CO2 and even the volatile allyl and butyl azide reacted successfully. The results reported in Table 4 show that all final products were obtained in excellent to almost quantitative yield.
Table 4: One-pot MW-assisted synthesis of a set of urea derivatives.a
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-274-i17.svg?max-width=637&scale=1.0)
|
||||
| entry | R–Br | R–NH2 | product | yieldb (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-274-i18.svg?max-width=637&scale=1.0)
17 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-274-i19.svg?max-width=637&scale=1.0)
22 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-274-i20.svg?max-width=637&scale=1.0)
27 |
98 |
| 2 | 17 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-274-i21.svg?max-width=637&scale=1.0)
23 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-274-i22.svg?max-width=637&scale=1.0)
28 |
98 |
| 3 | 17 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-274-i23.svg?max-width=637&scale=1.0)
24 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-274-i24.svg?max-width=637&scale=1.0)
29 |
98 |
| 4 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-274-i25.svg?max-width=637&scale=1.0)
18 |
22 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-274-i26.svg?max-width=637&scale=1.0)
10 |
97 |
| 5 | 18 | 23 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-274-i27.svg?max-width=637&scale=1.0)
30 |
94 |
| 6 | 18 | 24 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-274-i28.svg?max-width=637&scale=1.0)
31 |
98 |
| 7 | 18 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-274-i29.svg?max-width=637&scale=1.0)
25 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-274-i30.svg?max-width=637&scale=1.0)
32 |
98 |
| 8 | 18 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-274-i31.svg?max-width=637&scale=1.0)
26 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-274-i32.svg?max-width=637&scale=1.0)
33 |
98 |
| 9 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-274-i33.svg?max-width=637&scale=1.0)
19 |
22 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-274-i34.svg?max-width=637&scale=1.0)
34 |
89 |
| 10 | 19 | 23 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-274-i35.svg?max-width=637&scale=1.0)
35 |
89 |
| 11 | 19 | 24 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-274-i36.svg?max-width=637&scale=1.0)
36 |
88 |
| 12c |
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-274-i37.svg?max-width=637&scale=1.0)
20 |
3 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-9-274-i38.svg?max-width=637&scale=1.0)
37 |
85 |
| 13c |
![[Graphic 38]](/bjoc/content/inline/1860-5397-9-274-i39.svg?max-width=637&scale=1.0)
21 |
3 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-9-274-i40.svg?max-width=637&scale=1.0)
38 |
83 |
aReactions were carried out in a MW reactor: alkyl bromide, NaN3 (2 equiv), MeCN, 95 °C, 3 h ; then PS-PPh2 (1.5 equiv), CO2 (14.5 bar), amine (2 equiv), at 50 °C 1.5 h then 70 °C 3 h. bIsolated yield. cR–N3 was synthesized in DMF and MeCN was then added.
Conclusion
In conclusion, a MW-assisted, one-pot sequential protocol for the synthesis of urea derivatives from alkyl bromides has been described. This study has proven that in acetonitrile under high CO2 pressure the Staudinger–aza-Wittig reaction in presence of PS-PPh2 is strongly promoted. Excellent results have been obtained under MW irradiation in a closed vessel also with gaseous reagents. The optimized procedure benefited from the use of a quasi-stoichiometric amount of PS-PPh2 and can be applied for the efficient, safe, rapid, and cost-effective production of urea derivative libraries.
Experimental
All chemicals were purchased from Sigma-Aldrich (solvents from Carlo Erba SpA) and used without further purification. Solid diphenylphosphino-polystyrene (PS-PPh2) was purchased from Novabiochem® (Cas-No: 39319-11-4, loading ca. 1.2 mmol/g). Reactions were monitored by TLC on Merck 60 F254 (0.25 mm) plates, which were visualized by UV inspection and/or by heating after a spraying with 5% H2SO4 in ethanol or phosphomolybdic acid. MW-promoted reactions were carried out in a SynthWave (Milestone, Italy). NMR spectra were recorded on a Bruker Avance 300 (300 MHz and 75 MHz for 1H and 13C, respectively) at 25 °C; chemical shifts were calibrated to the residual proton and carbon resonances of the solvents: CDCl3 (δH = 7.26, δC = 77.16) or CD3OD (δH = 3.31, δC = 49.00). GC–MS analyses were performed in a GC Agilent 6890 (Agilent Technologies, USA) that was fitted with a mass detector Agilent Network 5973, using a 30 m long capillary column, i.d. of 0.25 mm and film thickness 0.25 μm. GC conditions were: injection split 1:20, injector temperature 250 °C, detector temperature 280 °C. Gas carrier: helium (1.2 mL/min), temperature program: from 70 °C (2 min) to 300 °C at 5 °C/min. HRMS was determined using MALDI-TOF mass spectra (Bruker Ultraflex TOF mass spectrometer).
General procedures
Representative procedure for alkyl isocyanate synthesis from alkyl azide: PS-PPh2 (0.477 mmol) was added to a solution of alkyl azide (0.318 mmol) in MeCN (1.5 mL). The mixture was irradiated by MW for 1.5 h at 50 °C (average power 70 W) under CO2 (14.5 bar) and magnetic stirring. After the reaction, the mixture was filtered on a cartridge. When the reaction was performed in a Parr reactor, PS-PPh2 (6.36 mmol) was added to a solution of alkyl azide (3.18 mmol) in MeCN (80 mL). The solution was heated for 2 h at 50 °C under CO2 (14.5 bar). After the reaction, the mixture was filtered on a cartridge.
Representative procedure for urea synthesis from alkyl isocyanate: The amine (0.636 mmol) was added to a solution of alkyl isocyanate (0.318 mmol) in MeCN (1.5 mL). The solution was irradiated by MW for 3 h at 70 °C (average power 200 W) under N2 (2 bar) and magnetic stirring. The solvent was then evaporated under vacuum, the residue was dissolved in MeOH and Dowex® 50WX8-200 was added. The mixture was stirred at rt for 15 min. The mixture was then filtered on paper and the solvent was evaporated under vacuum.
Representative “multi-pot” procedure for the synthesis of urea derivatives: NaN3 (0.477 mmol) was added to a solution of alkyl bromide (0.318 mmol) in MeCN (1.5 mL). The mixture was irradiated by MW for 3 h at 95 °C (average power 240 W) under N2 (2 bar) and magnetic stirring. After the reaction, the mixture was cooled to rt, filtered on paper, and PS-PPh2 (0.477 mmol) and amine (0.636 mmol) were sequentially added. The mixture was irradiated by MW for 1.5 h at 50 °C (average power 70 W) and 3 h at 70 °C (average power 200 W) under CO2 (14.5 bar) and magnetic stirring. Then, the mixture was filtered on a cartridge to remove the polymer-bound diphenylphosphine oxide. The solvent was then evaporated under vacuum, the residue was dissolved in MeOH and Dowex® 50WX8-200 was added. The mixture was stirred at rt for 15 min. Finally, the mixture was filtered on paper and the solvent was evaporated under vacuum. When the reaction was performed in a Parr reactor, PS-PPh2 (6.36 mmol) was added to a solution of alkyl azide (3.18 mmol) and amine (6.36 mmol) in MeCN (80 mL). The solution was heated 3 h at 70 °C under CO2 (14.5 bar). The mixture was filtered on a cartridge to remove the polymer-bound diphenylphosphine oxide and the residual polymer-bound diphenylphosphine. The solvent was then evaporated under vacuum, the residue was dissolved in MeOH and Dowex® 50WX8-200 was added. The mixture was stirred at rt for 15 min. Finally, the mixture was filtered on paper and the solvent was evaporated under vacuum.
Supporting Information
| Supporting Information File 1: Detailed analytical data of the prepared compounds and a collection of NMR spectra. | ||
| Format: PDF | Size: 2.2 MB | Download |
References
-
Delebecq, E.; Pascault, J.-P.; Boutevin, B.; Ganachaud, F. Chem. Rev. 2013, 113, 80–118. doi:10.1021/cr300195n
Return to citation in text: [1] -
Decicco, C. S.; Seng, J. L.; Kennedy, K. E.; Covington, M. B.; Welch, P. K.; Arner, E. C.; Magolda, R. L.; Nelson, D. J. Bioorg. Med. Chem. Lett. 1997, 7, 2331–2336. doi:10.1016/S0960-894X(97)00182-0
Return to citation in text: [1] -
Wurtz, A. Justus Liebigs Ann. Chem. 1849, 71, 326–342. doi:10.1002/jlac.18490710308
Return to citation in text: [1] -
Saunders, J. H.; Slocombe, R. J. Chem. Rev. 1948, 43, 203–218. doi:10.1021/cr60135a001
Return to citation in text: [1] -
Ozaki, S. Chem. Rev. 1972, 72, 457–496. doi:10.1021/cr60279a002
Return to citation in text: [1] -
Nowick, J. S.; Powell, N. A.; Nguyen, T. M.; Noronha, G. J. Org. Chem. 1992, 57, 7364–7366. doi:10.1021/jo00052a069
Return to citation in text: [1] -
Nowick, J. S.; Holmes, D. L.; Noronha, G.; Smith, E. M.; Nguyen, T. M.; Huang, S.-L. J. Org. Chem. 1996, 61, 3929–3934. doi:10.1021/jo960038n
Return to citation in text: [1] -
Twitchett, H. J. Chem. Soc. Rev. 1974, 3, 209–230. doi:10.1039/cs9740300209
Return to citation in text: [1] -
Kreye, O.; Mutlu, H.; Meier, M. A. R. Green Chem. 2013, 15, 1431–1455. doi:10.1039/c3gc40440d
Return to citation in text: [1] -
Lebel, H.; Leogane, O. Org. Lett. 2006, 8, 5717–5720. doi:10.1021/ol0622920
Return to citation in text: [1] -
Marinescu, L.; Thinggaard, J.; Thomsen, I. B.; Bols, M. J. Org. Chem. 2003, 68, 9453–9455. doi:10.1021/jo035163v
Return to citation in text: [1] -
Liu, P.; Wang, Z.; Hu, X. Eur. J. Org. Chem. 2012, 1994–2000. doi:10.1002/ejoc.201101784
Return to citation in text: [1] -
Dubé, P.; Nathel, N. F. F.; Vetelino, M.; Couturier, M.; Abossafy, C. L.; Pichette, S.; Jorgensen, M. L.; Hardink, M. Org. Lett. 2009, 11, 5622–5625. doi:10.1021/ol9023387
Return to citation in text: [1] -
Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; de los Santos, J. M. Tetrahedron 2007, 63, 523–575. doi:10.1016/j.tet.2006.09.048
Return to citation in text: [1] -
Eguchi, S. ARKIVOC 2005, No. ii, 98–119. doi:10.3998/ark.5550190.0006.208
Return to citation in text: [1] -
Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188–5240. doi:10.1002/anie.200400657
Return to citation in text: [1] -
Ayesa, S.; Samuelsson, B.; Classon, B. Synlett 2008, 97–99. doi:10.1055/s-2007-990927
Return to citation in text: [1] -
Hayashi, Y.; Itoh, T.; Nagae, N.; Ohkubo, M.; Ishikawa, H. Synlett 2008, 1565–1570. doi:10.1055/s-2008-1077789
Return to citation in text: [1] -
Mahdav, H.; Amani, J. Tetrahedron Lett. 2009, 50, 5923–5926. doi:10.1016/j.tetlet.2009.08.041
Return to citation in text: [1] -
Friant-Michel, P.; Marsura, A.; Kovacs, J.; Pintér, I.; Rivail, J.-L. THEOCHEM 1997, 395–396, 61–69. doi:10.1016/S0166-1280(97)00092-4
Return to citation in text: [1] -
Lindsley, C. W.; Zhao, Z.; Newton, R. C.; Leister, W. H.; Strauss, K. A. Tetrahedron Lett. 2002, 43, 4467–4470. doi:10.1016/S0040-4039(02)00827-4
Return to citation in text: [1] -
Lévêque, J.-M.; Cravotto, G. Chimia 2006, 60, 313–320. doi:10.2533/000942906777836255
Return to citation in text: [1] -
Cravotto, G.; Cintas, P. The Combined Use of Microwaves and Ultrasound. In Methods and Practice, Microwaves in Organic Synthesis, 3rd ed.; De La Hoz, A.; Loupy, A., Eds.; Springer: U.S.A., 2012.
Return to citation in text: [1] -
Cravotto, G.; Cintas, P. Chem.–Eur. J. 2007, 13, 1902–1909. doi:10.1002/chem.200601845
Return to citation in text: [1] -
Cintas, P.; Carnaroglio, D.; Rinaldi, L.; Cravotto, G. Chim. Oggi 2012, 30 (3), 33–35.
Return to citation in text: [1] -
Morschhäuser, R.; Krull, M.; Kayser, C.; Boberski, C.; Bierbaum, R.; Püschner, P. A.; Glasnov, T. N.; Kappe, C. O. Green Process. Synth. 2012, 1, 281–290. doi:10.1515/gps-2012-0032
Return to citation in text: [1] -
Kaval, N.; Dehaen, W.; Kappe, C. O.; Van der Eycken, E. Org. Biomol. Chem. 2004, 2, 154–156. doi:10.1039/b315150f
Return to citation in text: [1] -
Will, H.; Scholz, P.; Ondruschka, B. Chem. Ing. Tech. 2002, 74, 1057–1067. doi:10.1002/1522-2640(20020815)74:8<1057::AID-CITE1057>3.0.CO;2-3
Return to citation in text: [1] -
Tanner, D. D.; Kandanarachchi, P.; Ding, Q.; Shao, Q. H.; Vizitiu, D.; Franz, J. A. Energy Fuels 2001, 15, 197–204. doi:10.1021/ef000167d
Return to citation in text: [1] -
Zhang, X.; Lee, C. S.-M.; Mingos, D. M. P.; Hayward, D. O. Catal. Lett. 2003, 88, 129–139. doi:10.1023/A:1024049403422
Return to citation in text: [1] -
Zhang, X.; Hayward, D. O.; Mingos, D. M. P. Catal. Lett. 2003, 88, 33–38. doi:10.1023/A:1023530715368
Return to citation in text: [1] -
Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365–2387. doi:10.1021/cr068357u
Return to citation in text: [1] -
Yagodkin, A.; Löschcke, K.; Weisell, J.; Azhayev, A. Tetrahedron 2010, 66, 2210–2221. doi:10.1016/j.tet.2010.01.017
Return to citation in text: [1] -
Molina, P.; Aller, E.; Lorenzo, L.; López-Cremades, P.; Rioja, I.; Ubeda, A.; Terencio, M. C.; Alcaraz, M. J. J. Med. Chem. 2001, 44, 1011–1014. doi:10.1021/jm000997g
Return to citation in text: [1] -
Porwanski, S.; Kryczka, B.; Marsura, A. Tetrahedron Lett. 2002, 43, 8441–8443. doi:10.1016/S0040-4039(02)02101-9
Return to citation in text: [1] [2] -
Rao, H. S. P.; Siva, P. Synth. Commun. 1994, 24, 549–555. doi:10.1080/00397919408011505
Return to citation in text: [1] -
Fringuelli, F.; Pizzo, F.; Vaccaro, L. Synthesis 2000, 646–650. doi:10.1055/s-2000-6389
Return to citation in text: [1] -
Aime, S.; Gianolio, E.; Palmisano, P.; Robaldo, B.; Barge, A.; Boffa, L.; Cravotto, G. Org. Biomol. Chem. 2006, 4, 1124–1130. doi:10.1039/b517068k
Return to citation in text: [1] -
Cintas, P.; Cravotto, G.; Calcio Gaudino, E.; Orio, L.; Boffa, L. Catal. Sci. Technol. 2012, 2, 85–87. doi:10.1039/c1cy00378j
Return to citation in text: [1] -
Zhao, L.-J.; He, H. S.; Shi, M.; Toy, P. H. J. Comb. Chem. 2004, 6, 680–683. doi:10.1021/cc049917a
Return to citation in text: [1]
| 39. | Cintas, P.; Cravotto, G.; Calcio Gaudino, E.; Orio, L.; Boffa, L. Catal. Sci. Technol. 2012, 2, 85–87. doi:10.1039/c1cy00378j |
| 33. | Yagodkin, A.; Löschcke, K.; Weisell, J.; Azhayev, A. Tetrahedron 2010, 66, 2210–2221. doi:10.1016/j.tet.2010.01.017 |
| 34. | Molina, P.; Aller, E.; Lorenzo, L.; López-Cremades, P.; Rioja, I.; Ubeda, A.; Terencio, M. C.; Alcaraz, M. J. J. Med. Chem. 2001, 44, 1011–1014. doi:10.1021/jm000997g |
| 35. | Porwanski, S.; Kryczka, B.; Marsura, A. Tetrahedron Lett. 2002, 43, 8441–8443. doi:10.1016/S0040-4039(02)02101-9 |
| 36. | Rao, H. S. P.; Siva, P. Synth. Commun. 1994, 24, 549–555. doi:10.1080/00397919408011505 |
| 37. | Fringuelli, F.; Pizzo, F.; Vaccaro, L. Synthesis 2000, 646–650. doi:10.1055/s-2000-6389 |
| 38. | Aime, S.; Gianolio, E.; Palmisano, P.; Robaldo, B.; Barge, A.; Boffa, L.; Cravotto, G. Org. Biomol. Chem. 2006, 4, 1124–1130. doi:10.1039/b517068k |
| 35. | Porwanski, S.; Kryczka, B.; Marsura, A. Tetrahedron Lett. 2002, 43, 8441–8443. doi:10.1016/S0040-4039(02)02101-9 |
| 1. | Delebecq, E.; Pascault, J.-P.; Boutevin, B.; Ganachaud, F. Chem. Rev. 2013, 113, 80–118. doi:10.1021/cr300195n |
| 6. | Nowick, J. S.; Powell, N. A.; Nguyen, T. M.; Noronha, G. J. Org. Chem. 1992, 57, 7364–7366. doi:10.1021/jo00052a069 |
| 7. | Nowick, J. S.; Holmes, D. L.; Noronha, G.; Smith, E. M.; Nguyen, T. M.; Huang, S.-L. J. Org. Chem. 1996, 61, 3929–3934. doi:10.1021/jo960038n |
| 27. | Kaval, N.; Dehaen, W.; Kappe, C. O.; Van der Eycken, E. Org. Biomol. Chem. 2004, 2, 154–156. doi:10.1039/b315150f |
| 28. | Will, H.; Scholz, P.; Ondruschka, B. Chem. Ing. Tech. 2002, 74, 1057–1067. doi:10.1002/1522-2640(20020815)74:8<1057::AID-CITE1057>3.0.CO;2-3 |
| 29. | Tanner, D. D.; Kandanarachchi, P.; Ding, Q.; Shao, Q. H.; Vizitiu, D.; Franz, J. A. Energy Fuels 2001, 15, 197–204. doi:10.1021/ef000167d |
| 30. | Zhang, X.; Lee, C. S.-M.; Mingos, D. M. P.; Hayward, D. O. Catal. Lett. 2003, 88, 129–139. doi:10.1023/A:1024049403422 |
| 31. | Zhang, X.; Hayward, D. O.; Mingos, D. M. P. Catal. Lett. 2003, 88, 33–38. doi:10.1023/A:1023530715368 |
| 4. | Saunders, J. H.; Slocombe, R. J. Chem. Rev. 1948, 43, 203–218. doi:10.1021/cr60135a001 |
| 5. | Ozaki, S. Chem. Rev. 1972, 72, 457–496. doi:10.1021/cr60279a002 |
| 32. | Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365–2387. doi:10.1021/cr068357u |
| 3. | Wurtz, A. Justus Liebigs Ann. Chem. 1849, 71, 326–342. doi:10.1002/jlac.18490710308 |
| 24. | Cravotto, G.; Cintas, P. Chem.–Eur. J. 2007, 13, 1902–1909. doi:10.1002/chem.200601845 |
| 25. | Cintas, P.; Carnaroglio, D.; Rinaldi, L.; Cravotto, G. Chim. Oggi 2012, 30 (3), 33–35. |
| 2. | Decicco, C. S.; Seng, J. L.; Kennedy, K. E.; Covington, M. B.; Welch, P. K.; Arner, E. C.; Magolda, R. L.; Nelson, D. J. Bioorg. Med. Chem. Lett. 1997, 7, 2331–2336. doi:10.1016/S0960-894X(97)00182-0 |
| 26. | Morschhäuser, R.; Krull, M.; Kayser, C.; Boberski, C.; Bierbaum, R.; Püschner, P. A.; Glasnov, T. N.; Kappe, C. O. Green Process. Synth. 2012, 1, 281–290. doi:10.1515/gps-2012-0032 |
| 14. | Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; de los Santos, J. M. Tetrahedron 2007, 63, 523–575. doi:10.1016/j.tet.2006.09.048 |
| 15. | Eguchi, S. ARKIVOC 2005, No. ii, 98–119. doi:10.3998/ark.5550190.0006.208 |
| 16. | Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188–5240. doi:10.1002/anie.200400657 |
| 17. | Ayesa, S.; Samuelsson, B.; Classon, B. Synlett 2008, 97–99. doi:10.1055/s-2007-990927 |
| 18. | Hayashi, Y.; Itoh, T.; Nagae, N.; Ohkubo, M.; Ishikawa, H. Synlett 2008, 1565–1570. doi:10.1055/s-2008-1077789 |
| 19. | Mahdav, H.; Amani, J. Tetrahedron Lett. 2009, 50, 5923–5926. doi:10.1016/j.tetlet.2009.08.041 |
| 21. | Lindsley, C. W.; Zhao, Z.; Newton, R. C.; Leister, W. H.; Strauss, K. A. Tetrahedron Lett. 2002, 43, 4467–4470. doi:10.1016/S0040-4039(02)00827-4 |
| 10. | Lebel, H.; Leogane, O. Org. Lett. 2006, 8, 5717–5720. doi:10.1021/ol0622920 |
| 11. | Marinescu, L.; Thinggaard, J.; Thomsen, I. B.; Bols, M. J. Org. Chem. 2003, 68, 9453–9455. doi:10.1021/jo035163v |
| 12. | Liu, P.; Wang, Z.; Hu, X. Eur. J. Org. Chem. 2012, 1994–2000. doi:10.1002/ejoc.201101784 |
| 13. | Dubé, P.; Nathel, N. F. F.; Vetelino, M.; Couturier, M.; Abossafy, C. L.; Pichette, S.; Jorgensen, M. L.; Hardink, M. Org. Lett. 2009, 11, 5622–5625. doi:10.1021/ol9023387 |
| 22. | Lévêque, J.-M.; Cravotto, G. Chimia 2006, 60, 313–320. doi:10.2533/000942906777836255 |
| 23. | Cravotto, G.; Cintas, P. The Combined Use of Microwaves and Ultrasound. In Methods and Practice, Microwaves in Organic Synthesis, 3rd ed.; De La Hoz, A.; Loupy, A., Eds.; Springer: U.S.A., 2012. |
| 9. | Kreye, O.; Mutlu, H.; Meier, M. A. R. Green Chem. 2013, 15, 1431–1455. doi:10.1039/c3gc40440d |
| 40. | Zhao, L.-J.; He, H. S.; Shi, M.; Toy, P. H. J. Comb. Chem. 2004, 6, 680–683. doi:10.1021/cc049917a |
| 20. | Friant-Michel, P.; Marsura, A.; Kovacs, J.; Pintér, I.; Rivail, J.-L. THEOCHEM 1997, 395–396, 61–69. doi:10.1016/S0166-1280(97)00092-4 |
© 2013 Carnaroglio et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)