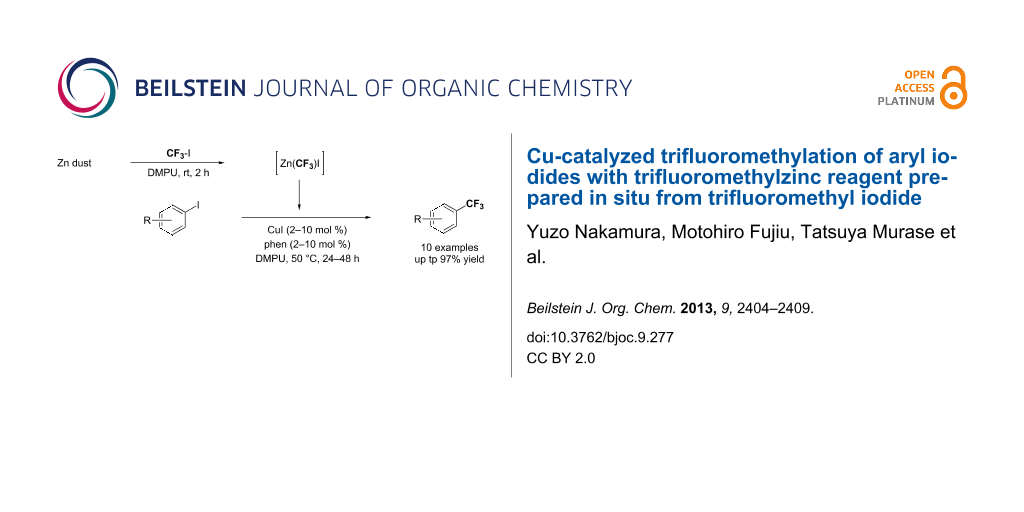
Abstract
The trifluoromethylation of aryl iodides catalyzed by copper(I) salt with trifluoromethylzinc reagent prepared in situ from trifluoromethyl iodide and Zn dust was accomplished. The catalytic reactions proceeded under mild reaction conditions, providing the corresponding aromatic trifluoromethylated products in moderate to high yields. The advantage of this method is that additives such as metal fluoride (MF), which are indispensable to activate silyl groups for transmetallation in the corresponding reactions catalyzed by copper salt by using the Ruppert–Prakash reagents (CF3SiR3), are not required.

Graphical Abstract
Introduction
Organo-fluorine compounds have received considerable attention in the fields of biomedical chemistry, agrochemistry, and organic material science due to their unique chemical, biological, and physical properties [1-6]. Particularly, trifluoromethylated compounds can be widely employed as one of the most effective analogues of bioactive compounds, because the trifluoromethyl group enhances the metabolic stability, lipophilicity, and bioavailability of these compounds [7]. As a result, trifluoromethylated compounds have been efficiently synthesized by both building-block methods which employ trifluoromethylated substrates and direct methods which employ trifluoromethyl reagents [7-9]. However, a nucleophilic trifluoromethylation by trifluoromethyl organometallic reagents such as lithium and magnesium, which are widely utilized in non-fluorine organic synthesis, cannot be used. These trifluoromethyl metal reagents are generally too unstable to prepare even at low temperature because of facile α-fluoro elimination generating the singlet difluoromethylene (:CF2) [10]. In contrast, trifluoromethylsilyl counterparts, so-called Ruppert–Prakash reagents (CF3SiR3), are highly stable, but reactive in the presence of fluoride, and hence the most versatile nucleophilic trifluoromethyl reagents [11,12]. The trifluoromethylzinc reagent (Zn(CF3)Br·2DMF), a stable solid, can also be used for the trifluoromethylation of aryl iodides, while stoichiometric amounts of copper(I) bromide are required to afford the more reactive trifluoromethyl copper (CuCF3) species by transmetallation [13,14]. However, these reagents are generally prepared from trifluoromethyl bromide (CF3Br), whose production is now prohibited because of the ozone depleting effect [15]. On the other hand, the trifluoromethylzinc reagent (Zn(CF3)I) formed in situ from trifluoromethyl iodide (CF3I) as an alternative trifluoromethyl source is utilized for trifluoromethylation reactions [10,16]. The preparation of the reagent followed by the reactions, however, requires ultrasonic irradiation and thus lacks reproducibility [16]. Therefore, the direct and reproducible preparation of the trifluoromethylzinc reagent and its application to trifluoromethylation reactions pose a particular challenge. Recently, Daugulis and co-workers reported the trifluoromethylation of aryl iodide catalyzed by copper(I) chloride with Zn(CF3)2 prepared in situ from TMP2Zn and fluoroform (CHF3), but only one substrate was investigated to provide the trifluoromethylated product only in a moderate yield [17]. As described above, much of the area of catalytic trifluoromethylations with trifluoromethylzinc reagent has not been explored yet, compared to the area of catalytic trifluoromethylations with the Ruppert–Prakash reagents [7,11,12,18-21]. Herein, we report the trifluoromethylations of aryl iodides catalyzed by copper(I) salt with trifluoromethylzinc reagent prepared in situ from CF3I and Zu dust. The trifluoromethylated aromatic products are privileged skeletal key compounds in pharmaceutical science as shown in Mefloquine (Lariam®), Fluoxetine (Prozac®), Leflunomide (Arava®), Celecoxib (Celebrex®), Bicalutamide (Casodex®), Aprepitant (Emend®), and Nilutamide (Nilandron®).
Results and Discussion
The preparation of the trifluoromethylzinc reagent Zn(CF3)I was initially examined in the context of the in situ Cu-catalyzed trifluoromethylation of aryl iodide 1 under various conditions. The results of the reaction are summarized in Table 1. After Zn(CF3)I was prepared in situ by the treatment of CF3I (ca. 5 equiv) with Zn dust (2 equiv) [22] in various solvents at room temperature for 2 hours, the reactions were explored by adding a catalytic amount of copper(I) salt and 1,10-phenanthroline (phen) [18-21] followed by aryl iodide 1a. In this catalytic system, solvents showed significant effects on the preparation and catalytic reactivity of Zn(CF3)I. No reaction was observed in less polar solvents such as toluene (Table 1, entry 1). Even in THF, CH3CN, DMSO, and NMP, the desired product 2a was barely or not at all obtained (Table 1, entries 2–5). Formation of Zn(CF3)I and reaction in DMF were found to proceed only with 44% yield (Table 1, entry 6). Replacement of DMF with DMPU provided the trifluoromethylated product 2a in 70% yield, along with the undesired pentafluoroethylated product 3a in 9% yield (Table 1, entry 7).
Table 1: Copper-catalyzed trifluoromethylation of aryl iodide 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-277-i3.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | Cu cat. | (X, Y) | T | % yield (2a/3a)a |
|---|---|---|---|---|---|
| 1 | toluene | CuI | 10, 20 | 50 | n.r. |
| 2 | THF | CuI | 10, 20 | 50 | n.r. |
| 3 | CH3CN | CuI | 10, 20 | 50 | 1/1 |
| 4 | DMSO | CuI | 10, 20 | 50 | 2/0 |
| 5 | NMP | CuI | 10, 20 | 50 | n.r. |
| 6 | DMF | CuI | 10, 20 | 50 | 44/8 |
| 7 | DMPU | CuI | 10, 20 | 50 | 70/9 |
| 8 | DMPU | CuI | 10, 10 | 50 | 93/7 |
| 9 | DMPU | CuI | 10, 10 | rt | 61/2 |
| 10 | DMPU | CuI | 10, 10 | 40 | 67/5 |
| 11 | DMPU | CuI | 10, 10 | 65 | 77/10 |
| 12 | DMPU | CuCl | 10, 10 | 50 | 92/8 |
| 13 | DMPU | CuTC | 10, 10 | 50 | 92/6 |
| 14 | DMPU | CuI | 2, 2 | 50 | 95/2 |
aYields were determined by 19F NMR analysis by using benzotrifluoride as an internal standard.
The product 3a should be derived from CuCF2CF3 generated by an insertion of difluoromethylene (:CF2) decomposed from CuCF3 into CuCF3 [23]. Decreasing the loading of phen, the yield of product 2a was further increased to exceed the level of 90% yield (Table 1, entry 8). The reaction was promoted even under the milder reaction conditions at room temperature (61% yield), but the highest yield was obtained at 50 °C (Table 1, entry 8 vs entries 9–11). A further change from CuI to CuCl and CuTC led to comparable results (Table 1, entries 12 and 13). With only a 2 mol % loading of both CuI and phen ligand, the yield of 2a was further increased to a higher level (95% yield) along with an increased selectivity with only 2% yield of 3a (Table 1, entry 14).
The ligand effect was further investigated in DMPU at 50 °C and phen was preferable to other diamine ligands (Table 2, entries 1–3 vs entry 4). Surprisingly, even in the absence of phen, it was found that the reaction smoothly proceeded to give a comparably high yield of product 2a (Table 2, entry 6). The reaction with a shorter reaction time of 2 hours indicated that the phen ligand slightly accelerated the reaction by the coordination to CuCF3 species, when compared to the reactions performed without the ligand (Table 2, entry 5 (78% yield) vs entry 6 (68% yield)). In the absence of CuI, no coupling product was obtained even in the presence of phen (Table 2, entries 7 and 8).
Table 2: Effect of ligands in copper-catalyzed trifluoromethylation.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-277-i4.svg?max-width=637&scale=1.0)
|
|||
| entry | ligand | (X, Y) | % yield (2a/3a)a |
|---|---|---|---|
| 1 | TMEDA | 10, 10 | 89/3 |
| 2 | DMEDA | 10, 10 | 83/3 |
| 3 | bipy | 10, 10 | 86/9 |
| 4 | phen | 10, 10 | 93/7 |
| 5 | phen | 2, 2 | 95/2 (78/1)b |
| 6 | – | 2, 0 | 97/3 (68/1)b |
| 7 | – | 0, 0 | n.r. |
| 8 | phen | 0, 2 | n.r. |
aYields were determined by 19F NMR analysis by using benzotrifluoride as an internal standard. bValues in parentheses are yields obtained with a reaction time of 2 hours.
With the reaction conditions established in DMPU at 50 °C in the presence of a catalytic amount of CuI and phen, the scope and limitation of this method were evaluated. The results are shown in Figure 1. The use of the electron-deficient aryl iodides 1b–f bearing nitrile, nitro, formyl, and trifluoromethyl groups led to the corresponding products 2b–f in moderate to high yields. The reactions of heteroaryl iodides 1g–i were also catalyzed to provide the corresponding products 2g–i in good to excellent yields. In the case of 1h, the formation of a CF3 group occurred only at the position of iodide, and bromide remained intact during the course of reaction. It was found that an increase of the yield in the presence of the phen ligand depends on the particular substrate, while the yields were within the same range except for 2b and 2d. Unfortunately, aryl iodide 1j bearing the electron-donating methoxy substituent extremely decreased the reactivity, even when increasing the catalytic amounts of CuI and phen.
![[1860-5397-9-277-1]](/bjoc/content/figures/1860-5397-9-277-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Copper-catalyzed trifluoromethylation of various aryl iodides. Yields were determined by 19F NMR analysis by using benzotrifluoride as an internal standard. Values in parentheses are yields obtained under the reaction conditions without phen. Conditions: CF3I (2.5 × X equiv) and Zn dust (X equiv) in DMPU, then CuI (Y mol %), phen (Y mol %) and 1 (1 equiv) at 50 °C for t hours. aX = 4, Y = 2, t = 48. bX = 2, Y = 10, t = 24. cIsolated yields: 2c, 70%; 2h, 90%.
Figure 1: Copper-catalyzed trifluoromethylation of various aryl iodides. Yields were determined by 19F NMR an...
In order to gain an insight into each step of the catalytic trifluoromethylation with a trifluoromethylzinc reagent, a 19F NMR analysis in DMF and DMPU was performed (Scheme 1). At the initial stage, Zn(CF3)I which readily causes a Schlenk equilibrium with Zn(CF3)2 and ZnI2 [14,24] was prepared in situ from CF3I and Zn dust in 60–80% yields in both solvents. The addition of CuI (0.2 equiv) to a DMPU solution of Zn(CF3)I led to the transmetallation of the CF3 group from zinc to copper even at room temperature. Two singlet peaks of the cuprate species, [Cu(CF3)I]− (−29.7 ppm) and [Cu(CF3)2]− (−31.9 ppm) were observed in 12% and 1% yields, respectively [23,25,26]. By replacing DMPU with DMF as a solvent, the transmetallation was found to be less efficient than in DMPU. Moreover, the inactive copper species [Cu(CF3)4] − (−34.8 ppm) [27] was obtained. The neutral CuCF3 species (−26.3 ppm), which formed by the direct cupration of fluoroform in DMF [28] and was active even with aryl iodides bearing electron-donating substituents such as 1j [28-30], was not observed in both solvents. Thus, the addition of aryl iodide 1a led to the formation of the trifluoromethyl coupling product 2a (−59.8 ppm) even in the absence of phen, involving the consumption of the cuprates [Cu(CF3)I] − and [Cu(CF3)2] −. The use of DMF as a solvent led to a gradual increase of the peak assigned as inactive [Cu(CF3)4] − during the course of the reaction.
![[1860-5397-9-277-i1]](/bjoc/content/inline/1860-5397-9-277-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Observation of CuCF3 species in 19F NMR spectrum. aEquivalents based on Zn(CF3)I. bYields based on CuI.
Scheme 1: Observation of CuCF3 species in 19F NMR spectrum. aEquivalents based on Zn(CF3)I. bYields based on ...
The mechanism of the coupling reaction can thus be visualized by the following catalytic cycle (Scheme 2). At the first step, the transmetallation of the CF3 group to CuI from Zn(CF3)I or Zn(CF3)2 affords the active cuprate species, [Cu(CF3)X]− (X = I, CF3). Subsequently, the oxidative addition to the cuprate of aryl iodide 1, and the reductive elimination gives the desired cross-coupling product 2 together with the formation of ZnI2.
![[1860-5397-9-277-i2]](/bjoc/content/inline/1860-5397-9-277-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism of copper-catalyzed trifluoromethylation.
Scheme 2: Proposed mechanism of copper-catalyzed trifluoromethylation.
Conclusion
In summary, we succeeded in the aromatic trifluoromethylation catalyzed by copper(I) salt with a trifluoromethylzinc reagent prepared in situ from trifluoromethyl iodide and Zn dust in DMPU. The catalytic reaction proceeded to provide moderate to high yields and a high selectivity of the trifluoromethylated product under mild reaction conditions. The advantage of this catalytic reaction is that additives such as metal fluoride (MF), which are indispensable to activate silyl substituents for the transmetallation in the corresponding catalytic reactions by using the Ruppert–Prakash reagents, are not necessary. Additionally, with some substrates, the reaction conditions without a ligand led to higher yields than reaction conditions with a ligand such as 1,10-phenanthroline. Further studies on highly efficient trifluoromethylation and difluoromethylation reactions with trifluoromethylzinc reagents are under way.
Experimental
Typical procedure for copper-catalyzed trifluoromethylation of aryl iodide
To the suspension of zinc powder (without activation, 65.4 mg, 1.0 mmol, Aldrich 99.995% purity) in DMPU (0.5 mL), trifluoromethyl iodide (ca. 2.5 mmol, sufficiently dissolved in the solution) was added at room temperature under argon atmosphere. After the solution was stirred for 2 h at room temperature, CuI (1.9 mg, 0.01 mmol, 2 mol %), 1.10-phenanthroline (1.8 mg, 0.01 mmol, 2 mol %), and then aryl iodide 1a (138.0 mg, 0.5 mmol) were added. The reaction mixture was stirred at 50 °C for 24 h. After cooling to room temperature, the yield of product 2a was determined by 19F NMR analysis by using benzotrifluoride (BTF) as an internal standard. Except for 2c, all trifluoromethylated products 2 exhibited the same 1H, 13C, and 19F NMR spectra as reported before [14,17,29,31-36].
2,4-Bis(trifluoromethyl)benzonitrile (2c)
To the suspension of zinc powder (without activation, 65.4 mg, 1.0 mmol, Aldrich 99.995 % purity) in DMPU (0.5 mL), trifluoromethyl iodide (ca. 2.5 mmol, sufficiently dissolved in the solution) was added at room temperature under argon atmosphere. The solution was stirred for 2 h, and CuI (9.5 mg, 0.05 mmol, 10 mol %) and 4-iodo-2-(trifluoromethyl)benzonitrile (1c, 148.5 mg, 0.5 mmol) were added. The reaction mixture was stirred at 50 °C for 24 h. The reaction mixture was quenched with H2O (5 mL), and then Et2O (5 mL) was added. After filtration over celite, the organic layer was separated, and the aqueous layer was extracted with Et2O (5 mL × 3). The combined organic layer was washed with brine (10 mL), dried over Na2SO4, and evaporated. The resulting crude product was purified by silica gel column chromatography (pentane/Et2O 9:1) to give the product 2c (83 mg, 70% yield) as a colorless liquid. 1H NMR (300 MHz, CDCl3) δ 8.06 (s, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.98 (d, J = 8.2 Hz, 1H); 13C NMR (400 MHz, CDCl3) δ 135.5, 135.0 (q, JCF = 34.4 Hz), 134.0 (q, JCF = 33.5 Hz), 129.3 (q, JCF = 3.6 Hz), 124.1–123.9 (m), 122.3 (q, JCF = 271.9 Hz), 121.6 (q, JCF = 272.7 Hz), 114.1, 113.9; 19F NMR (282 MHz, CDCl3) δ −62.2 (s, 3F), −63.6 (s, 3F); HRMS–ESITOF (m/z): [M − H]− calcd for C9H2F6N, 238.0091; found, 238.0086; FTIR (neat, cm−1) 2238, 1344, 1146, 1279, 1082.
Observation of trifluoromethylcopper species in 19F NMR spectrum
To the suspension of zinc powder (without activation, 32.7 mg, 0.5 mmol, Aldrich 99.995% purity) in DMF or DMPU (0.75 mL), trifluoromethyl iodide (ca. 1.25 mmol, 2.5 equiv, sufficiently dissolved to the solution) was added at room temperature under argon atmosphere. After the solution was stirred for 2 h at room temperature, the remaining trifluoromethyl iodide was removed by bubbling argon through the solution for 15 min. To the solution was added CuI (19.0 mg, 0.1 mmol) at room temperature. After the reaction mixture was stirred for 5 min, the generation of cuprate species was monitored by 19F NMR analysis by using benzotrifluoride (10 μL, 0.0814 mmol) as an internal standard and sealed capillary filled with benzene-d6 for signal lock under argon atmosphere at room temperature.
Acknowledgements
This research was partly supported by a grant program “Creation of Advanced Catalytic Transformation for the Sustainable Manufacturing at Low Energy, Low Environmental Load (ACT-C)” from the Japan Science and Technology Agency (JST). We thank TOSOH F-TECH, INC. for donating the trifluoromethyl iodide.
References
-
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351
Return to citation in text: [1] -
Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, U.K., 2006. doi:10.1002/9780470988589
Return to citation in text: [1] -
Hiyama, T. Organofluorine Compounds: Chemistry and Applications; Springer-Verlag: Berlin, 2000. doi:10.1007/978-3-662-04164-2
Return to citation in text: [1] -
Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, U.K., 2009.
Return to citation in text: [1] -
Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470–477. doi:10.1038/nature10108
Return to citation in text: [1] -
Jin, Z.; Hammond, G. B.; Xu, B. Aldrichimica Acta 2012, 45, 67–83.
Return to citation in text: [1] -
Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293
Return to citation in text: [1] [2] [3] -
Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a
Return to citation in text: [1] -
Mikami, K.; Itoh, Y.; Yamanaka, M. Chem. Rev. 2004, 104, 1–16. doi:10.1021/cr030685w
Return to citation in text: [1] -
Burton, D. J.; Yang, Z.-Y. Tetrahedron 1992, 48, 189–275. doi:10.1016/S0040-4020(01)88139-4
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8
Return to citation in text: [1] [2] -
Naumann, D.; Tyrra, W.; Kock, B.; Rudolph, W.; Wilkes, B. J. Fluorine Chem. 1994, 67, 91–93. doi:10.1016/0022-1139(93)02937-A
Return to citation in text: [1] -
Kremlev, M. M.; Tyrra, W.; Mushta, A. I.; Naumann, D.; Yagupolskii, Y. L. J. Fluorine Chem. 2010, 131, 212–216. doi:10.1016/j.jfluchem.2009.10.011
Return to citation in text: [1] [2] [3] -
Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859
Return to citation in text: [1] -
Kitazume, T.; Ishikawa, N. J. Am. Chem. Soc. 1985, 107, 5186–5191. doi:10.1021/ja00304a026
Return to citation in text: [1] [2] -
Popov, I.; Lindeman, S.; Daugulis, O. J. Am. Chem. Soc. 2011, 133, 9286–9289. doi:10.1021/ja2041942
Return to citation in text: [1] [2] -
Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k
Return to citation in text: [1] [2] -
Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679–1681. doi:10.1126/science.1190524
Return to citation in text: [1] [2] -
Chu, L.; Qing, F.-L. J. Am. Chem. Soc. 2012, 134, 1298–1304. doi:10.1021/ja209992w
Return to citation in text: [1] [2] -
Jiang, X.; Chu, L.; Qing, F.-L. J. Org. Chem. 2012, 77, 1251–1257. doi:10.1021/jo202566h
Return to citation in text: [1] [2] -
Aldrich: Lot# MKBK3648V, powder <150 µm, 99.995% trace metals basis.
Return to citation in text: [1] -
Wiemers, D. M.; Burton, D. J. J. Am. Chem. Soc. 1986, 108, 832–834. doi:10.1021/ja00264a043
Return to citation in text: [1] [2] -
Francèse, C.; Tordeux, M.; Wakselman, C. Tetrahedron Lett. 1988, 29, 1029–1030. doi:10.1016/0040-4039(88)85326-7
Return to citation in text: [1] -
Hu, M.; Ni, C.; Hu, J. J. Am. Chem. Soc. 2012, 134, 15257–15260. doi:10.1021/ja307058c
Return to citation in text: [1] -
Kütt, A.; Movchun, V.; Rodima, T.; Dansauer, T.; Rusanov, E. B.; Leito, I.; Kaljurand, I.; Koppel, J.; Pihl, V.; Koppel, I.; Ovsjannikov, G.; Toom, L.; Mishima, M.; Medebielle, M.; Lork, E.; Röschenthaler, G.-V.; Koppel, I. A.; Kolomeitsev, A. A. J. Org. Chem. 2008, 73, 2607–2620. doi:10.1021/jo702513w
Return to citation in text: [1] -
Naumann, D.; Roy, T.; Tebbe, K.-F.; Crump, W. Angew. Chem., Int. Ed. Engl. 1993, 32, 1482–1483. doi:10.1002/anie.199314821
Return to citation in text: [1] -
Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026
Return to citation in text: [1] [2] -
Morimoto, H.; Tsubogo, T.; Litvinas, N. D.; Hartwig, J. F. Angew. Chem., Int. Ed. 2011, 50, 3793–3798. doi:10.1002/anie.201100633
Return to citation in text: [1] [2] -
Serizawa, H.; Aikawa, K.; Mikami, K. Chem.–Eur. J., in press.
Return to citation in text: [1] -
Anbarasan, P.; Neumann, H.; Beller, M. Chem.–Eur. J. 2011, 17, 4217–4222. doi:10.1002/chem.201003388
Return to citation in text: [1] -
Kondratenko, N. V.; Vechirko, E. P.; Yagupolskii, L. M. Synthesis 1980, 932–933. doi:10.1055/s-1980-29276
Return to citation in text: [1] -
Kremlev, M. M.; Mushta, A. I.; Tyrra, W.; Yagupolskii, Y. L.; Naumann, D.; Möller, A. J. Fluorine Chem. 2012, 133, 67–71. doi:10.1016/j.jfluchem.2011.07.025
Return to citation in text: [1] -
Ye, Y.; Sanford, M. S. J. Am. Chem. Soc. 2012, 134, 9034–9037. doi:10.1021/ja301553c
Return to citation in text: [1] -
Knauber, T.; Arikan, F.; Röschenthaler, G.-V.; Gooßen, L. J. Chem.–Eur. J. 2011, 17, 2689–2697. doi:10.1002/chem.201002749
Return to citation in text: [1] -
Lumma, W. C., Jr.; Hartman, R. D.; Saari, W. S.; Engelhardt, E. L.; Hirschmann, R.; Clineschmidt, B. V.; Torchiana, M. L.; Stone, C. A. J. Med. Chem. 1978, 21, 536–542. doi:10.1021/jm00204a007
Return to citation in text: [1]
| 28. | Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026 |
| 29. | Morimoto, H.; Tsubogo, T.; Litvinas, N. D.; Hartwig, J. F. Angew. Chem., Int. Ed. 2011, 50, 3793–3798. doi:10.1002/anie.201100633 |
| 30. | Serizawa, H.; Aikawa, K.; Mikami, K. Chem.–Eur. J., in press. |
| 27. | Naumann, D.; Roy, T.; Tebbe, K.-F.; Crump, W. Angew. Chem., Int. Ed. Engl. 1993, 32, 1482–1483. doi:10.1002/anie.199314821 |
| 28. | Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026 |
| 1. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351 |
| 2. | Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, U.K., 2006. doi:10.1002/9780470988589 |
| 3. | Hiyama, T. Organofluorine Compounds: Chemistry and Applications; Springer-Verlag: Berlin, 2000. doi:10.1007/978-3-662-04164-2 |
| 4. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, U.K., 2009. |
| 5. | Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470–477. doi:10.1038/nature10108 |
| 6. | Jin, Z.; Hammond, G. B.; Xu, B. Aldrichimica Acta 2012, 45, 67–83. |
| 11. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 12. | Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8 |
| 14. | Kremlev, M. M.; Tyrra, W.; Mushta, A. I.; Naumann, D.; Yagupolskii, Y. L. J. Fluorine Chem. 2010, 131, 212–216. doi:10.1016/j.jfluchem.2009.10.011 |
| 24. | Francèse, C.; Tordeux, M.; Wakselman, C. Tetrahedron Lett. 1988, 29, 1029–1030. doi:10.1016/0040-4039(88)85326-7 |
| 10. | Burton, D. J.; Yang, Z.-Y. Tetrahedron 1992, 48, 189–275. doi:10.1016/S0040-4020(01)88139-4 |
| 23. | Wiemers, D. M.; Burton, D. J. J. Am. Chem. Soc. 1986, 108, 832–834. doi:10.1021/ja00264a043 |
| 25. | Hu, M.; Ni, C.; Hu, J. J. Am. Chem. Soc. 2012, 134, 15257–15260. doi:10.1021/ja307058c |
| 26. | Kütt, A.; Movchun, V.; Rodima, T.; Dansauer, T.; Rusanov, E. B.; Leito, I.; Kaljurand, I.; Koppel, J.; Pihl, V.; Koppel, I.; Ovsjannikov, G.; Toom, L.; Mishima, M.; Medebielle, M.; Lork, E.; Röschenthaler, G.-V.; Koppel, I. A.; Kolomeitsev, A. A. J. Org. Chem. 2008, 73, 2607–2620. doi:10.1021/jo702513w |
| 7. | Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 8. | Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a |
| 9. | Mikami, K.; Itoh, Y.; Yamanaka, M. Chem. Rev. 2004, 104, 1–16. doi:10.1021/cr030685w |
| 18. | Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k |
| 19. | Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679–1681. doi:10.1126/science.1190524 |
| 20. | Chu, L.; Qing, F.-L. J. Am. Chem. Soc. 2012, 134, 1298–1304. doi:10.1021/ja209992w |
| 21. | Jiang, X.; Chu, L.; Qing, F.-L. J. Org. Chem. 2012, 77, 1251–1257. doi:10.1021/jo202566h |
| 7. | Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 23. | Wiemers, D. M.; Burton, D. J. J. Am. Chem. Soc. 1986, 108, 832–834. doi:10.1021/ja00264a043 |
| 16. | Kitazume, T.; Ishikawa, N. J. Am. Chem. Soc. 1985, 107, 5186–5191. doi:10.1021/ja00304a026 |
| 7. | Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 11. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 12. | Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8 |
| 18. | Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k |
| 19. | Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679–1681. doi:10.1126/science.1190524 |
| 20. | Chu, L.; Qing, F.-L. J. Am. Chem. Soc. 2012, 134, 1298–1304. doi:10.1021/ja209992w |
| 21. | Jiang, X.; Chu, L.; Qing, F.-L. J. Org. Chem. 2012, 77, 1251–1257. doi:10.1021/jo202566h |
| 10. | Burton, D. J.; Yang, Z.-Y. Tetrahedron 1992, 48, 189–275. doi:10.1016/S0040-4020(01)88139-4 |
| 16. | Kitazume, T.; Ishikawa, N. J. Am. Chem. Soc. 1985, 107, 5186–5191. doi:10.1021/ja00304a026 |
| 15. | Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859 |
| 14. | Kremlev, M. M.; Tyrra, W.; Mushta, A. I.; Naumann, D.; Yagupolskii, Y. L. J. Fluorine Chem. 2010, 131, 212–216. doi:10.1016/j.jfluchem.2009.10.011 |
| 17. | Popov, I.; Lindeman, S.; Daugulis, O. J. Am. Chem. Soc. 2011, 133, 9286–9289. doi:10.1021/ja2041942 |
| 29. | Morimoto, H.; Tsubogo, T.; Litvinas, N. D.; Hartwig, J. F. Angew. Chem., Int. Ed. 2011, 50, 3793–3798. doi:10.1002/anie.201100633 |
| 31. | Anbarasan, P.; Neumann, H.; Beller, M. Chem.–Eur. J. 2011, 17, 4217–4222. doi:10.1002/chem.201003388 |
| 32. | Kondratenko, N. V.; Vechirko, E. P.; Yagupolskii, L. M. Synthesis 1980, 932–933. doi:10.1055/s-1980-29276 |
| 33. | Kremlev, M. M.; Mushta, A. I.; Tyrra, W.; Yagupolskii, Y. L.; Naumann, D.; Möller, A. J. Fluorine Chem. 2012, 133, 67–71. doi:10.1016/j.jfluchem.2011.07.025 |
| 34. | Ye, Y.; Sanford, M. S. J. Am. Chem. Soc. 2012, 134, 9034–9037. doi:10.1021/ja301553c |
| 35. | Knauber, T.; Arikan, F.; Röschenthaler, G.-V.; Gooßen, L. J. Chem.–Eur. J. 2011, 17, 2689–2697. doi:10.1002/chem.201002749 |
| 36. | Lumma, W. C., Jr.; Hartman, R. D.; Saari, W. S.; Engelhardt, E. L.; Hirschmann, R.; Clineschmidt, B. V.; Torchiana, M. L.; Stone, C. A. J. Med. Chem. 1978, 21, 536–542. doi:10.1021/jm00204a007 |
| 13. | Naumann, D.; Tyrra, W.; Kock, B.; Rudolph, W.; Wilkes, B. J. Fluorine Chem. 1994, 67, 91–93. doi:10.1016/0022-1139(93)02937-A |
| 14. | Kremlev, M. M.; Tyrra, W.; Mushta, A. I.; Naumann, D.; Yagupolskii, Y. L. J. Fluorine Chem. 2010, 131, 212–216. doi:10.1016/j.jfluchem.2009.10.011 |
| 17. | Popov, I.; Lindeman, S.; Daugulis, O. J. Am. Chem. Soc. 2011, 133, 9286–9289. doi:10.1021/ja2041942 |
© 2013 Nakamura et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)