Abstract
The trifluoromethylation of iodoarenes was accomplished by use of a 2-trifluoromethylbenzimidazoline derivative as the trifluoromethylating reagent and a catalytic amount of Cu(I) in the presence of 2,2'-bipyridyl as the ligand. Through a mechanistic study, we found that the oxidative addition of the iodoarene to the Cu(I)–CF3 species is the rate-determining step.

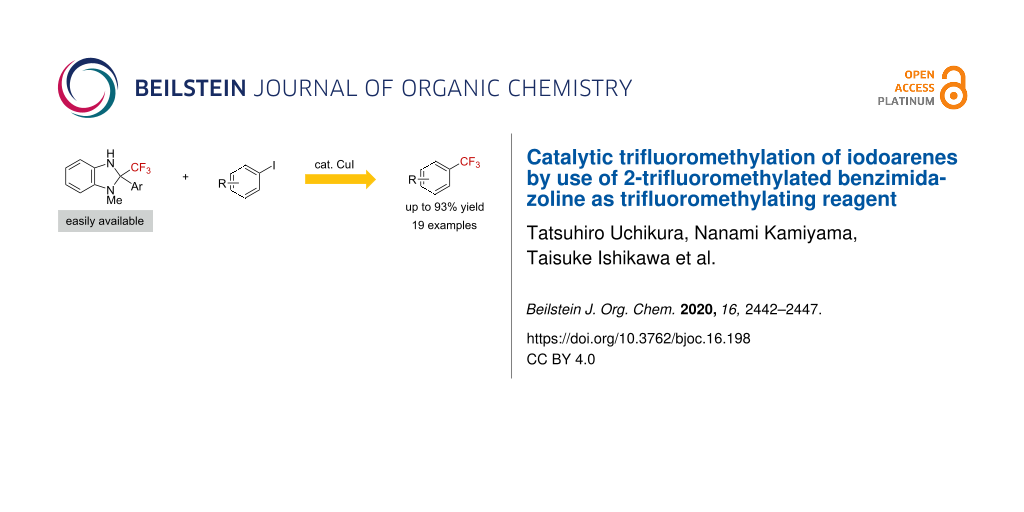
Graphical Abstract
Introduction
The introduction of a trifluoromethyl group is one of the most attractive reactions in drug discovery [1,2]. In the past decade, trifluoromethylation reactions of aryl halides in the presence of transition-metal complexes were reported [3-21]. CuCF3 is a useful species for the trifluoromethylation of aryl halides and there are a number of precursors of CuCF3 for trifluoromethylation reactions. In contrast, the catalytic generation of CuCF3 was less investigated [15-21]. For example, R3SiCF3, a fluoral derivative, and trifluoroacetates were employed as precursors of CuCF3 species for the catalytic trifluoromethylation of iodoarenes (Figure 1a) and the development of novel types of trifluoromethylating reagents is still desired. We have recently reported the trifluoromethylation of iodoarenes by use of 2-aryl-2-trifluoromethylbenzimidazoline as the trifluoromethylating reagent in the presence of 3 equiv of a copper salt [22]. The benzimidazoline derivatives could be readily prepared from relatively cheap materials, namely, trifluoromethylacetophenone and phenylenediamine derivatives (Figure 1b). Herein we report a catalytic trifluoromethylation of iodoarenes by use of benzimidazoline derivatives in the presence of a catalytic amount of copper salts and a bipyridyl ligand (Figure 1c).
![[1860-5397-16-198-1]](/bjoc/content/figures/1860-5397-16-198-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Trifluoromethylation of aryl halides.
Figure 1: Trifluoromethylation of aryl halides.
Results and Discussion
We first investigated the reaction conditions by use of p-iodonitrobenzene (1a) and 2-phenyl-2-trifluoromethyl-1-methylbenzimidazoline (2) (Table 1). Using 3 equiv of CuI gave 4-trifluoromethyl-1-nitrobenzene (3a) quantitatively, as we had reported previously (Table 1, entry 1) [22]. Decreasing the CuI catalyst loading to 20 mol % lowered the yield of 3a even at a high temperature (Table 1, entries 2 and 3). In order to stabilize the copper(I) catalyst, a bipyridyl ligand was employed in the reaction [23]. By this, the trifluoromethylation proceeded with a catalytic amount of CuI in the presence of 0.8 equiv of 2,2’-bipyridyl to furnish product 3a in 80% yield (Table 1, entry 4). However, smaller amounts of CuI (10 mol %) and 2,2’-bipyridyl (20 mol %) gave compound 3a in lower yields (Table 1, entries 5 and 6). Benzonitrile was the solvent of choice (Table 1, entries 7 and 8). Replacing CuI by CuBr or CuCl gave similar results (Table 1, entries 9 and 10). However, we selected the most stable and readily available CuI as the catalyst for further investigations. Thus, the trifluoromethylation of p-iodonitrobenzene proceeded by use of 2-phenyl-2-trifluoromethyl-1-methylbenzimidazoline in the presence of catalytic amounts of CuI and 2,2’-bipyridyl as ligand.
Table 1: Screening for reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-198-i1.svg?max-width=637&scale=1.0)
|
|||||
| entry | X | Y | solvent | temp. | yieldb |
|---|---|---|---|---|---|
| 1 | 3 | 0 | PhCN | 60 °C | quant |
| 2 | 0.2 | 0 | PhCN | 60 °C | 36% |
| 3 | 0.2 | 0 | PhCN | 90 °C | 40% |
| 4 | 0.2 | 0.8 | PhCN | 90 °C | 80% |
| 5 | 0.1 | 0.8 | PhCN | 90 °C | 59% |
| 6 | 0.2 | 0.2 | PhCN | 90 °C | 63% |
| 7 | 0.2 | 0.8 | MeCN | 90 °C | 32% |
| 8 | 0.2 | 0.8 | DMF | 90 °C | 30% |
| 9c | 0.2 | 0.8 | PhCN | 90 °C | 72% |
| 10d | 0.2 | 0.8 | PhCN | 90 °C | 79% |
aPerformed with 2a (0.050 mmol) and 1a (0.10 mmol) in solvent (1.0 mL). bDetermined by 19F NMR spectroscopy (benzotrifluoride was used as the internal standard). cCuBr was used instead of CuI. dCuCl was used instead of CuI.
We next screened for 2,2’-bipyridyl ligands to be used with CuI (Table 2). 4,4’-Dimethyl-2,2’-bipyridyl (4b) gave product 3a in a higher yield than 2,2’-bipyridyl (4a), whereas 5,5’- and 6,6’-dimethylbipyridyl (4c and 4d) were not effective. From these results, the substituents at 4,4’-positions were expected to be beneficial for the reaction, and other 4,4’-substituted bipyridyl ligands were investigated. We found that 4,4’-di-tert-butyl-2,2’-bipyridyl (4e) afforded the best result (91% yield). On the other hand, 2,2'-bipyridyl ligands bearing electron-donating (–OMe, 4f) and -withdrawing (–CF3, 4g) groups furnished 3a in low yields. Moreover, other electron-withdrawing ligands, i.e., phenanthroline derivatives (4h and 4i), and electron-donating ligands such as 2,2’-biimidazole (4j) and tetramethylethylenediamine (4k) gave inferior results.
Table 2: Screening for diamine ligandsa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-198-i2.svg?max-width=637&scale=1.0)
|
|||
| ligand | yieldb | ligand | yieldb |
|---|---|---|---|
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-198-i3.svg?max-width=637&scale=1.0)
4a |
80% |
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-198-i4.svg?max-width=637&scale=1.0)
4g |
56% |
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-198-i5.svg?max-width=637&scale=1.0)
4b |
82% |
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-198-i6.svg?max-width=637&scale=1.0)
4h |
64% |
![[Graphic 7]](/bjoc/content/inline/1860-5397-16-198-i7.svg?max-width=637&scale=1.0)
4c |
72% |
![[Graphic 8]](/bjoc/content/inline/1860-5397-16-198-i8.svg?max-width=637&scale=1.0)
4i |
57% |
![[Graphic 9]](/bjoc/content/inline/1860-5397-16-198-i9.svg?max-width=637&scale=1.0)
4d |
19% |
![[Graphic 10]](/bjoc/content/inline/1860-5397-16-198-i10.svg?max-width=637&scale=1.0)
4j |
trace |
![[Graphic 11]](/bjoc/content/inline/1860-5397-16-198-i11.svg?max-width=637&scale=1.0)
4e |
91% |
![[Graphic 12]](/bjoc/content/inline/1860-5397-16-198-i12.svg?max-width=637&scale=1.0)
4k |
26% |
![[Graphic 13]](/bjoc/content/inline/1860-5397-16-198-i13.svg?max-width=637&scale=1.0)
4f |
73% | – | 40% |
aPerformed with 2a (0.050 mmol) and 1a (0.10 mmol) in solvent (1.0 mL). bDetermined by 19F NMR spectroscopy (benzotrifluoride was used as the internal standard).
We next screened for the generality of the reaction towards various substrates under the optimized conditions (Figure 2). Electron-deficient aryl iodides were well tolerated furnishing the corresponding trifluoromethylation products in high yields. Among the tested nitrophenyl derivatives, p- and o-nitrophenyliodide gave the products in highest yields. In contrast, m-iodonitrobenzene afforded the trifluoromethylated product in a decreased yield of 35% due to the higher electron density of the meta-position compared to the ortho- and para-positions. Iodoarenes bearing other electron-withdrawing substituents, such as p-cyano, p-acetyl, and p-methoxycarbonyl, were also suitable and gave products 3d–f in moderate to high yields. Furthermore, the presence of a formyl group was also tolerated in the reaction, and p-formyltrifluoromethylbenzene (3g) was obtained in 40% yield. However, the electron-rich substrate 2-methoxyiodobenzene (1h) gave product 3h in only a modest yield (30%).
![[1860-5397-16-198-2]](/bjoc/content/figures/1860-5397-16-198-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Scope of aryl iodides. Yields determined by 19F NMR spectroscopy and used ligand is given in parenthesis.
Figure 2: Scope of aryl iodides. Yields determined by 19F NMR spectroscopy and used ligand is given in parent...
Heteroaryl iodides were also suitable substrates (Figure 3). 2-Iodopyridine (5a) gave the expected trifluoromethylation product 6a in 80% yield. 2-Iodoquinoline (5b) and 1-iodoisoquinoline (5c) were also suitable substrates to furnish desired products 6b and 6c in high yields. Furthermore, iodopyrazine was applicable to furnish 6d in 63% yield.
![[1860-5397-16-198-3]](/bjoc/content/figures/1860-5397-16-198-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Scope of heteroaryl iodides. Yields determined by 19F NMR spectroscopy and used ligand is given in parenthesis.
Figure 3: Scope of heteroaryl iodides. Yields determined by 19F NMR spectroscopy and used ligand is given in ...
Finally, a mechanistic study of the reaction was carried out. First, the active species of the reaction was investigated by NMR analysis. The generation of Cu(I)–CF3 was observed by mixing benzimidazoline 2 and CuI in EtCN at 90 °C (Figure S1, in Supporting Information File 1). Therefore, CuI and 2 generated CuCF3 species as the active species for the trifluoromethylation [24].
Then, the dependence of the conversion on the reaction time was estimated (Figure 4) and no induction period was observed. Although benzimidazoline 2 was completely consumed after 24 h, the yield of the trifluoromethylation product continued to increase up to 48 h. This suggests that product generation proceeded slower than the cleavage of the C–CF3 bond of benzimidazoline.
![[1860-5397-16-198-4]](/bjoc/content/figures/1860-5397-16-198-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Time course of the trifluoromethylation reaction.
Figure 4: Time course of the trifluoromethylation reaction.
We propose a mechanism for the reaction, as shown in Figure 5. The Cu(I)–CF3 species, generated through the reaction of benzimidazoline 2 with CuI under basic conditions, underwent an oxidative addition reaction with the aryl iodide to generate a Cu(III) complex. A subsequent reductive elimination furnished the trifluoromethylarene and Cu(I). Because an electron-donating ligand was more effective than an electron-deficient one (Table 2), and the reaction with benzimidazoline proceeded rapidly (Figure 4), the oxidative addition was suggested to be the rate-determining step.
![[1860-5397-16-198-5]](/bjoc/content/figures/1860-5397-16-198-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Proposed mechanism of the catalytic cycle.
Figure 5: Proposed mechanism of the catalytic cycle.
Conclusion
In conclusion, we have developed a catalytic trifluoromethylation of aryl iodides by using trifluoromethylated benzimidazoline derivatives. The mechanistic study revealed that the oxidative addition was the rate-determining step of this reaction. 2-Phenyl-2-trifluoromethyl-1-methylbenzimidazoline is a novel type of trifluoromethylating reagents that might be useful for organic synthesis.
Experimental
General procedure of trifluoromethylation: Aryl iodide 1 (0.1 mmol), 2 (56 mg, 0.2 mmol), CuI (3.8 mg, 0.02 mmol), 2,2’-bipyridyl (12.5 mg, 0.08 mmol), and potassium carbonate (55.6 mg, 0.4 mmol) were mixed in benzonitrile (1.0 mL), and the mixture heated to 90 °C. After 48 h, hexafluorobenzene was added as an internal standard and the mixture analyzed by 19F NMR spectroscopy for the calculation of the NMR yield. Then, the crude products were purified by preparative TLC to give pure products 3.
Supporting Information
| Supporting Information File 1: Details of screening experiments, synthetic procedures and characterization data of new compounds, and copies of spectra. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Alonso, C.; Martínez de Marigorta, E.; Rubiales, G.; Palacios, F. Chem. Rev. 2015, 115, 1847–1935. doi:10.1021/cr500368h
Return to citation in text: [1] -
Li, G.-b.; Zhang, C.; Song, C.; Ma, Y.-d. Beilstein J. Org. Chem. 2018, 14, 155–181. doi:10.3762/bjoc.14.11
Return to citation in text: [1] -
Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683–730. doi:10.1021/cr400473a
Return to citation in text: [1] -
Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k
Return to citation in text: [1] -
Zhang, C. Org. Biomol. Chem. 2014, 12, 6580. doi:10.1039/c4ob00671b
Return to citation in text: [1] -
Umemoto, T.; Zhang, B.; Zhu, T.; Zhou, X.; Zhang, P.; Hu, S.; Li, Y. J. Org. Chem. 2017, 82, 7708–7719. doi:10.1021/acs.joc.7b00669
Return to citation in text: [1] -
Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026
Return to citation in text: [1] -
Chen, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2013, 52, 11628–11631. doi:10.1002/anie.201306094
Return to citation in text: [1] -
Lin, X.; Hou, C.; Li, H.; Weng, Z. Chem. – Eur. J. 2016, 22, 2075–2084. doi:10.1002/chem.201504306
Return to citation in text: [1] -
Zhang, C.-P.; Wang, Z.-L.; Chen, Q.-Y.; Zhang, C.-T.; Gu, Y.-C.; Xiao, J.-C. Angew. Chem., Int. Ed. 2011, 50, 1896–1900. doi:10.1002/anie.201006823
Return to citation in text: [1] -
Li, X.; Zhao, J.; Zhang, L.; Hu, M.; Wang, L.; Hu, J. Org. Lett. 2015, 17, 298–301. doi:10.1021/ol5034018
Return to citation in text: [1] -
Negishi, K.; Aikawa, K.; Mikami, K. Eur. J. Org. Chem. 2016, 4099–4104. doi:10.1002/ejoc.201600711
Return to citation in text: [1] -
Chen, Q.-Y.; Wu, S.-W. J. Chem. Soc., Chem. Commun. 1989, 705–706. doi:10.1039/c39890000705
Return to citation in text: [1] [2] -
Kondo, H.; Oishi, M.; Fujikawa, K.; Amii, H. Adv. Synth. Catal. 2011, 353, 1247–1252. doi:10.1002/adsc.201000825
Return to citation in text: [1] [2] -
Knauber, T.; Arikan, F.; Röschenthaler, G.-V.; Gooßen, L. J. Chem. – Eur. J. 2011, 17, 2689–2697. doi:10.1002/chem.201002749
Return to citation in text: [1] [2] -
Li, Y.; Chen, T.; Wang, H.; Zhang, R.; Jin, K.; Wang, X.; Duan, C. Synlett 2011, 1713–1716. doi:10.1055/s-0030-1260930
Return to citation in text: [1] [2] -
Nakamura, Y.; Fujiu, M.; Murase, T.; Itoh, Y.; Serizawa, H.; Aikawa, K.; Mikami, K. Beilstein J. Org. Chem. 2013, 9, 2404–2409. doi:10.3762/bjoc.9.277
Return to citation in text: [1] [2] -
Aikawa, K.; Nakamura, Y.; Yokota, Y.; Toya, W.; Mikami, K. Chem. – Eur. J. 2015, 21, 96–100. doi:10.1002/chem.201405677
Return to citation in text: [1] [2] -
Shimizu, N.; Kondo, H.; Oishi, M.; Fujikawa, K.; Komada, K.; Amii, H. Org. Synth. 2016, 93, 147–162. doi:10.15227/orgsyn.093.0147
Return to citation in text: [1] [2] -
Miyagawa, M.; Ishikawa, T.; Shinkai, K.; Akiyama, T. J. Fluorine Chem. 2019, 219, 29–31. doi:10.1016/j.jfluchem.2018.12.006
Return to citation in text: [1] [2] -
The bipyridyl ligands stabilize the copper(I) catalyst for catalytic reaction (see ref. [6]).
Return to citation in text: [1] -
From the examination of an aryl group at the 2-position of benzimidazolines, electron-rich moieties were more reactive than electron-deficient ones (Table S5, see Supporting Information File 1). These results suggest that an oxidative addition step proceeds by nucleophilic process.
Return to citation in text: [1]
| 1. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 2. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 22. | Miyagawa, M.; Ishikawa, T.; Shinkai, K.; Akiyama, T. J. Fluorine Chem. 2019, 219, 29–31. doi:10.1016/j.jfluchem.2018.12.006 |
| 22. | Miyagawa, M.; Ishikawa, T.; Shinkai, K.; Akiyama, T. J. Fluorine Chem. 2019, 219, 29–31. doi:10.1016/j.jfluchem.2018.12.006 |
| 15. | Chen, Q.-Y.; Wu, S.-W. J. Chem. Soc., Chem. Commun. 1989, 705–706. doi:10.1039/c39890000705 |
| 16. | Kondo, H.; Oishi, M.; Fujikawa, K.; Amii, H. Adv. Synth. Catal. 2011, 353, 1247–1252. doi:10.1002/adsc.201000825 |
| 17. | Knauber, T.; Arikan, F.; Röschenthaler, G.-V.; Gooßen, L. J. Chem. – Eur. J. 2011, 17, 2689–2697. doi:10.1002/chem.201002749 |
| 18. | Li, Y.; Chen, T.; Wang, H.; Zhang, R.; Jin, K.; Wang, X.; Duan, C. Synlett 2011, 1713–1716. doi:10.1055/s-0030-1260930 |
| 19. | Nakamura, Y.; Fujiu, M.; Murase, T.; Itoh, Y.; Serizawa, H.; Aikawa, K.; Mikami, K. Beilstein J. Org. Chem. 2013, 9, 2404–2409. doi:10.3762/bjoc.9.277 |
| 20. | Aikawa, K.; Nakamura, Y.; Yokota, Y.; Toya, W.; Mikami, K. Chem. – Eur. J. 2015, 21, 96–100. doi:10.1002/chem.201405677 |
| 21. | Shimizu, N.; Kondo, H.; Oishi, M.; Fujikawa, K.; Komada, K.; Amii, H. Org. Synth. 2016, 93, 147–162. doi:10.15227/orgsyn.093.0147 |
| 3. | Alonso, C.; Martínez de Marigorta, E.; Rubiales, G.; Palacios, F. Chem. Rev. 2015, 115, 1847–1935. doi:10.1021/cr500368h |
| 4. | Li, G.-b.; Zhang, C.; Song, C.; Ma, Y.-d. Beilstein J. Org. Chem. 2018, 14, 155–181. doi:10.3762/bjoc.14.11 |
| 5. | Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683–730. doi:10.1021/cr400473a |
| 6. | Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k |
| 7. | Zhang, C. Org. Biomol. Chem. 2014, 12, 6580. doi:10.1039/c4ob00671b |
| 8. | Umemoto, T.; Zhang, B.; Zhu, T.; Zhou, X.; Zhang, P.; Hu, S.; Li, Y. J. Org. Chem. 2017, 82, 7708–7719. doi:10.1021/acs.joc.7b00669 |
| 9. | Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026 |
| 10. | Chen, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2013, 52, 11628–11631. doi:10.1002/anie.201306094 |
| 11. | Lin, X.; Hou, C.; Li, H.; Weng, Z. Chem. – Eur. J. 2016, 22, 2075–2084. doi:10.1002/chem.201504306 |
| 12. | Zhang, C.-P.; Wang, Z.-L.; Chen, Q.-Y.; Zhang, C.-T.; Gu, Y.-C.; Xiao, J.-C. Angew. Chem., Int. Ed. 2011, 50, 1896–1900. doi:10.1002/anie.201006823 |
| 13. | Li, X.; Zhao, J.; Zhang, L.; Hu, M.; Wang, L.; Hu, J. Org. Lett. 2015, 17, 298–301. doi:10.1021/ol5034018 |
| 14. | Negishi, K.; Aikawa, K.; Mikami, K. Eur. J. Org. Chem. 2016, 4099–4104. doi:10.1002/ejoc.201600711 |
| 15. | Chen, Q.-Y.; Wu, S.-W. J. Chem. Soc., Chem. Commun. 1989, 705–706. doi:10.1039/c39890000705 |
| 16. | Kondo, H.; Oishi, M.; Fujikawa, K.; Amii, H. Adv. Synth. Catal. 2011, 353, 1247–1252. doi:10.1002/adsc.201000825 |
| 17. | Knauber, T.; Arikan, F.; Röschenthaler, G.-V.; Gooßen, L. J. Chem. – Eur. J. 2011, 17, 2689–2697. doi:10.1002/chem.201002749 |
| 18. | Li, Y.; Chen, T.; Wang, H.; Zhang, R.; Jin, K.; Wang, X.; Duan, C. Synlett 2011, 1713–1716. doi:10.1055/s-0030-1260930 |
| 19. | Nakamura, Y.; Fujiu, M.; Murase, T.; Itoh, Y.; Serizawa, H.; Aikawa, K.; Mikami, K. Beilstein J. Org. Chem. 2013, 9, 2404–2409. doi:10.3762/bjoc.9.277 |
| 20. | Aikawa, K.; Nakamura, Y.; Yokota, Y.; Toya, W.; Mikami, K. Chem. – Eur. J. 2015, 21, 96–100. doi:10.1002/chem.201405677 |
| 21. | Shimizu, N.; Kondo, H.; Oishi, M.; Fujikawa, K.; Komada, K.; Amii, H. Org. Synth. 2016, 93, 147–162. doi:10.15227/orgsyn.093.0147 |
| 6. | Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k |
| 24. | From the examination of an aryl group at the 2-position of benzimidazolines, electron-rich moieties were more reactive than electron-deficient ones (Table S5, see Supporting Information File 1). These results suggest that an oxidative addition step proceeds by nucleophilic process. |
| 23. | The bipyridyl ligands stabilize the copper(I) catalyst for catalytic reaction (see ref. [6]). |
© 2020 Uchikura et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)