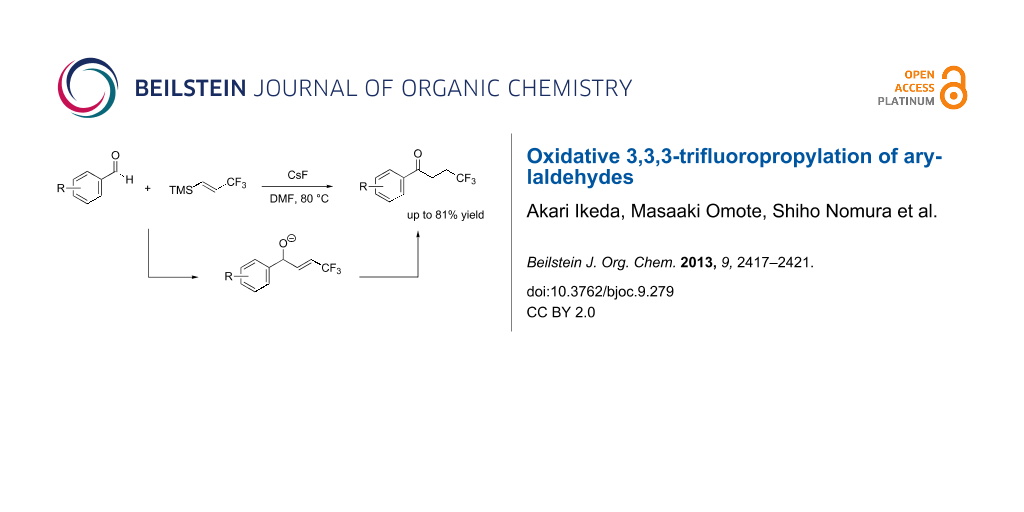
Abstract
A reaction between (E)-trimethyl(3,3,3-trifluoroprop-1-en-1-yl)silane (1) and arylaldehydes 2 was triggered by fluoride anions to afford aryl 3,3,3-trifluoropropyl ketones 3 in moderate to good yield. A mechanistic study of this reaction indicated that it occurred via an allyl alkoxide (4). A subsequent 1,3-proton shift of the benzylic proton of 4 forms 3. This reaction involves oxidative 3,3,3-trifluoropropylation of an arylaldehyde to afford 4,4,4-trifluoro-1-arylbutan-1-one.

Graphical Abstract
Introduction
Trifluoromethyl groups are an essential motif in pharmaceuticals, agricultural chemicals, and functional materials because trifluoromethylation of such chemicals often significantly improves their performance [1-6]. To date, the trifluoromethylation of carbonyl compounds [7-10] and aryl halides [11-13] has been extensively explored. On the other hand, the 2,2,2-trifluoroethylation and the 3,3,3-trifluoropropylation, which elongate the product by two and three carbon atoms, respectively, have not been explored yet. 2-Bromo-3,3,3-trifluoropropene [14-26] and 1,1,1,3,3-pentafluoropropane [27,28] have been used in place of (3,3,3-trifluoropropynyl)lithium, which can add to carbonyl compounds and couple with aryl halides through a zinc intermediate. For the 3,3,3-trifluoropropenyl synthon Yamazaki et al. reported the use of 2-(trifluoromethyl)-1-(phenylsulfenyl)vinyltrimethylsilane for the addition to aldehydes in the presence of fluoride anion [29]. Recently, Prakash et al. [30] reported the synthesis of β-trifluoromethylstyrenes through a Heck coupling reaction of aryl iodides with 1-iodo-3,3,3-trifluoropropane, allowing a direct introduction of 3,3,3-trifluoropropenyl groups to aromatic rings. Approximately at the same time, our research group demonstrated that (E)-trimethyl(3,3,3-trifluoroprop-1-en-1-yl)silane (1) effectively participated in a Hiyama cross-coupling reaction with aryl iodide to construct β-trifluoromethylstyrenes in good to excellent yield [31,32]. In the course of this study, we found that the reaction of 1 with benzaldehyde in the presence of fluoride anions afforded an allyl alcohol, which spontaneously isomerized to phenyl 3,3,3-trifluoropropyl ketone (3a). This reaction involves a one-pot synthesis of aryl 3,3,3-trifluoropropyl ketones from the corresponding arylaldehyde, representing the oxidative 3,3,3-propenylation of the arylaldehyde. Here, we report our study of this unusual reaction.
Results and Discussion
At the outset of our study, we attempted selective C–Si bond dissociation of 1 with several fluoride anion sources. The subsequently generated carbanion was trapped with benzaldehyde to generate 4,4,4-trifluoromethylallyl alcohol 4 (Table 1). We found that this reaction did not take place when either TiF4 in THF or CuF2/dppp in DMF was used (Table 1, entries 1 and 2). However, the reaction proceeded when two equivalents of CsF to arylaldehyde 2 were used in DMA at a temperature of 55 °C, affording 3a and 4a in 15% and 16% yield, respectively (Table 1, entry 3). In contrast, the reaction did not proceed in THF (Table 1, entry 4). The yield increased as the temperature was raised to 80 °C, but a further increase to 100 °C did not improve the yield of the reaction (Table 1, entries 5 and 6). Changing the solvent to DMF considerably shortened the reaction time. The added amount of CsF also influenced this reaction. The reaction conducted with four equivalents of CsF afforded 3a and 4a in 52% and 27% yield, respectively (Table 1, entry 8). However, a further increase of the amount of CsF did not improve the reaction yield (Table 1, entry 9). We thus determined that the conditions used in entry 8 of Table 1 were optimal. We explored the scope and limitation of this reaction by using these optimal conditions.
Table 1: Reaction of 1 with benzaldehyde in the presence of fluoride anions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-279-i3.svg?max-width=637&scale=1.0)
|
|||||||
| entry | F anion (equiv)b | 1 (equiv)b | solvent | temp (°C) | time (h) | yield (%)c,d | |
|---|---|---|---|---|---|---|---|
| 3a | 4a | ||||||
| 1 | TiF4 (2) | 2 | THF | 60 | 24 | – | – |
| 2 | CuF2 (1)/dppp (2) | 2 | DMF | 80 | 26 | – | – |
| 3 | CsF (2) | 1.5 | DMA | 55 | 24 | 15 | 16 |
| 4 | CsF (2) | 1.5 | THF | 55 | 24 | – | – |
| 5 | CsF (2) | 2 | DMA | 80 | 4 | 53 | – |
| 6 | CsF (2) | 2 | DMA | 100 | 4 | 47 | – |
| 7 | CsF (2) | 2 | DMF | 80 | 1 | 49 | 10 |
| 8 | CsF (4) | 2 | DMF | 80 | 1 | 52 (43) | 27 |
| 9 | CsF (6) | 2 | DMF | 80 | 1 | 38 | 5 |
aThe reaction was carried out with 2a (0.2 mmol) and a solvent (2.0 mL). bThe value in parentheses indicates equiv with respect to benzaldehyde (0.2 mmol). cNMR yields, which were calculated by integration of the 19F NMR signals of 3a and 4a relative to that of the internal standard of 1,4-bis(trifluoromethyl)benzene. dThe value in parentheses indicates isolated yield (%).
A variety of arylaldehydes participated in the reaction to give products in moderate to good yields (Table 2). The reactions with benzaldehyde derivatives with an electron-withdrawing group, such as chloro-, bromo-, fluoro- or trifluoromethyl, at the para-position proceeded to give 3b–e in moderate to good yields (Table 2, entries 1 to 4). The substitution with other electron-withdrawing groups such as methoxycarbonyl and cyano decreased the yield of the product, 3f and 3g were obtained in 38% and 46% yield, respectively (Table 2, entries 5 and 6). In contrast, the substitution of benzaldehyde with an electron-donating group slowed down the reaction substantially. p-Methyl and p-methoxybenzaldehydes gave 3h and 3i in 26% and 17% yield, respectively (Table 2, entries 7 and 8). meta-Substitution of benzaldehyde with a methoxy group gave 3j in 58% yield (Table 2, entry 9). These results are in accordance with the Hammett equation. ortho-Substitution of substrates decreased the yield of the products substantially, probably because of the steric hindrance introduced by the substituent (Table 2, entries 10 and 11). While the reaction could be applied to polycyclic and heterocyclic compounds, 3m and 3n were only obtained in low yield (Table 2, entries 12 and 13). An aliphatic aldehyde did not participate in the reaction (Table 2, entry 14).
Table 2: Investigation of reaction scope with optimal conditions.a
| entry | Ar–CHO | 3 | yield (%)b | 4 | yield (%)b |
|---|---|---|---|---|---|
| 1 | p-Cl-C6H4 | 3b | 72 | 4b | 8 |
| 2 | p-Br-C6H4 | 3c | 79 | 4c | 6 |
| 3 | p-F-C6H4 | 3d | 55 | 4d | 4 |
| 4 | p-CF3-C6H4 | 3e | 59 | 4e | 6 |
| 5 | p-CH3OC(O)-C6H4 | 3f | 38 | 4f | 6 |
| 6 | p-CN-C6H4 | 3g | 46 | 4g | 10 |
| 7 | p-CH3-C6H4 | 3h | 26 | 4h | 8 |
| 8 | p-CH3O-C6H4 | 3i | 17 | 4i | 16 |
| 9 | m-CH3O-C6H4 | 3j | 58 | 4j | 10 |
| 10 | o-CH3O-C6H4 | 3k | 12 | 4k | 35 |
| 11 | o-Cl-C6H4 | 3l | 16 | 4l | 16 |
| 12 | 2-naphthyl | 3m | 41 | 4m | 5 |
| 13 | 3-pyridyl | 3n | 31 | 4n | 12 |
| 14 | C6H5(CH2)2 | 3o | – | 3o | – |
aReactions were carried out with 1 (0.4 mmol), 2 (0.2 mmol) and CsF (0.8 mmol) in DMF (2.0 mL) at 80 °C. bNMR yields, which were calculated by integration of the 19F NMR signals 3 and 4 relative to that of the internal standard of 1,4-bis(trifluoromethyl)benzene.
During the course of this study, it became obvious that the reaction gave allyl alcohol 4 first, and then subsequent transformation of 4 into 3 occurred during the reaction process. Recently, both Cahard et al. [33,34] and Qing et al. [35] independently reported that the isomerization of a 4,4,4-trifluoromethylallyl alcohol substructure was promoted by a ruthenium catalyst to form a 3,3,3-trifluoropropylcarbonyl unit. We hypothesized that the formation of 3 would be promoted by inter- or intramolecular migration of the benzylic proton of 4 in the basic medium. To confirm the hypothesis, we conducted the reaction by using benzaldehyde-d to clarify whether or not proton migration was involved in the reaction sequence. When benzaldehyde-d was treated with 1 and CsF in DMF at 80 °C, the reaction gave a product in which deuterium was inserted on the α- and β-carbons of the carbonyl group with rates of 22% and 53%, respectively (Scheme 1, (1)). This result suggests that deuterium on the β-carbon should be incorporated by a 1,3-proton shift. In contrast, deuterium on the α-carbon should be inserted when the enolate intermediate, which is generated during the reaction, intermolecularly extracted the benzylic deuterium of 4. To further investigate the reaction mechanism, 4a was exposed to basic conditions to observe whether or not the isomerization of 4a to 3a would occur. Indeed, the treatment of 4a with a stoichiometric amount of DBU resulted in an almost quantitative conversion to 3a (Scheme 1, (2)).
![[1860-5397-9-279-i1]](/bjoc/content/inline/1860-5397-9-279-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Experiments to elucidate the reaction mechanism.
Scheme 1: Experiments to elucidate the reaction mechanism.
This suggests that the reaction provides 4 first. 4 subsequently isomerizes to form 3, which is an oxidative 3,3,3-trifluoropropylation of the arylaldehyde. These results allowed us to propose the reaction mechanism shown in Scheme 2, which includes the isomerization of 5 to 6 in the formation of 3. To support our proposed mechanism, a computational calculation was performed with Gaussian 03W at the B3LYP/6-31+G* level of theory to confirm that key intermediate 7 was involved in the isomerization process. The calculation indicated that intermediate 5 generated in the first step of the reaction was slightly more stable (0.417 kcal/mol) than 7. The slight energy difference between 5 and 7 indicates the viability of 7 in the reaction medium. The generation of 7 would facilitate the isomerization of 5 to 6.
![[1860-5397-9-279-i2]](/bjoc/content/inline/1860-5397-9-279-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The proposed reaction mechanism.
Scheme 2: The proposed reaction mechanism.
Conclusion
We reacted 1 with benzaldehyde in the presence of CsF to provide 3. We demonstrated that 4 was generated in the first step of the reaction, and then subsequently isomerized to 3 under basic conditions. This sequential reaction involves an oxidative 3,3,3-trifluoropropylation of the arylaldehyde to form a 3,3,3-trifluoropropylcarbonyl unit. A further refinement of the reaction conditions to enhance the compatibility of the reaction with substrates such as aliphatic aldehydes is in progress in our group.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data for all new compounds. | ||
| Format: PDF | Size: 337.4 KB | Download |
References
-
Tomaschenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293
Return to citation in text: [1] -
Yamazaki, T.; Taguchi, T.; Ojima, I. Unique Properties of Fluorine and Their Relevance to Medicinal Chemistry and Chemical Biology. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Wiley-Blackwell: Chichester, U.K., 2009; pp 3–46.
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Hiyama, T. In Organofluorine Chemistry, Principles, and Commercial Applications; Banks, R. E.; Smart, B. E.; Tatlow, C. J., Eds.; Springer: New York, 1994; pp 237–262.
Return to citation in text: [1] -
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] -
Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A. J. Am. Chem. Soc. 1989, 111, 393–395. doi:10.1021/ja00183a073
Return to citation in text: [1] -
Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011
Return to citation in text: [1] -
Ma, J.-A.; Cahard, D. J. Fluorine Chem. 2007, 128, 975–996. doi:10.1016/j.jfluchem.2007.04.026
Return to citation in text: [1] -
Kobayashi, Y.; Kumadaki, I. Tetrahedron Lett. 1969, 10, 4095–4096. doi:10.1016/S0040-4039(01)88624-X
Return to citation in text: [1] -
Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k
Return to citation in text: [1] -
Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679–1681. doi:10.1126/science.1190524
Return to citation in text: [1] -
Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/C39950000051
Return to citation in text: [1] -
Yamazaki, T.; Mizutani, K.; Kitazume, T. J. Org. Chem. 1995, 60, 6046–6056. doi:10.1021/jo00124a013
Return to citation in text: [1] -
Konno, T.; Chae, J.; Kanda, M.; Nagai, G.; Tamura, K.; Ishihara, T.; Yamanaka, H. Tetrahedron 2003, 59, 7571–7580. doi:10.1016/S0040-4020(03)01199-2
Return to citation in text: [1] -
Chen, Q.; Qiu, X.-L.; Qing, F.-L. J. Fluorine Chem. 2007, 128, 1182–1186. doi:10.1016/j.jfluchem.2007.02.010
Return to citation in text: [1] -
Jiang, Z.-X.; Qin, Y.-Y.; Qing, F.-L. J. Org. Chem. 2003, 68, 7544–7547. doi:10.1021/jo0344384
Return to citation in text: [1] -
Xiong, Z.; Qiu, X.-L.; Huang, Y.; Qing, F.-L. J. Fluorine Chem. 2011, 132, 166–174. doi:10.1016/j.jfluchem.2010.12.012
Return to citation in text: [1] -
Hanamoto, T.; Hakoshima, Y.; Egashira, M. Tetrahedron Lett. 2004, 45, 7573–7576. doi:10.1016/j.tetlet.2004.08.118
Return to citation in text: [1] -
Chen, X.-Y.; Qiu, X.-L.; Qing, F.-L. Tetrahedron 2008, 64, 2301–2306. doi:10.1016/j.tet.2008.01.021
Return to citation in text: [1] -
Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073–4076. doi:10.1021/ol048229x
Return to citation in text: [1] -
Watanabe, Y.; Yamazaki, T. J. Org. Chem. 2011, 76, 1957–1960. doi:10.1021/jo102503s
Return to citation in text: [1] -
Yamazaki, Y.; Yamamoto, T.; Ichihara, R. J. Org. Chem. 2006, 71, 6251–6253. doi:10.1021/jo060909l
Return to citation in text: [1] -
Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945–5948. doi:10.1016/j.tet.2009.05.087
Return to citation in text: [1] -
Konno, T.; Moriyasu, K.; Ishihara, T. Synthesis 2009, 1087–1094. doi:10.1055/s-0028-1087988
Return to citation in text: [1] -
Brisdon, A. K.; Crossley, I. R.; Pritchard, R. G.; Sadiq, G.; Warren, J. E. Organometallics 2003, 22, 5534–5542. doi:10.1021/om034146t
Return to citation in text: [1] -
Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 20, 2420–2421. doi:10.1039/b207979h
Return to citation in text: [1] -
Yamazaki, T.; Takita, K.; Ishikawa, N. J. Fluorine Chem. 1985, 30, 357–363. doi:10.1016/S0022-1139(00)84971-4
Return to citation in text: [1] -
Prakash, G. K. S.; Krishnan, H. S.; Jog, P. V.; Iyer, A. P.; Olah, G. A. Org. Lett. 2012, 14, 1146–1149. doi:10.1021/ol300076y
Return to citation in text: [1] -
Omote, M.; Tanaka, M.; Ikeda, A.; Nomura, S.; Tarui, A.; Sato, K.; Ando, A. Org. Lett. 2012, 14, 2286–2289. doi:10.1021/ol300670n
Return to citation in text: [1] -
Omote, M.; Tanaka, M.; Tanaka, M.; Ikeda, A.; Tarui, A.; Sato, K.; Ando, A. J. Org. Chem. 2013, 78, 6196–6201. doi:10.1021/jo400859s
Return to citation in text: [1] -
Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. Angew. Chem., Int. Ed. 2012, 51, 6467–6470. doi:10.1002/anie.201200827
Return to citation in text: [1] -
Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56–61. doi:10.1016/j.jfluchem.2013.01.004
Return to citation in text: [1] -
Qing, F.; Gao, W. Chin. J. Org. Chem. 2000, 20, 764–768.
http://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract333670.shtml
Return to citation in text: [1]
| 1. | Tomaschenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 2. | Yamazaki, T.; Taguchi, T.; Ojima, I. Unique Properties of Fluorine and Their Relevance to Medicinal Chemistry and Chemical Biology. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Wiley-Blackwell: Chichester, U.K., 2009; pp 3–46. |
| 3. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 4. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 5. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 6. | Hiyama, T. In Organofluorine Chemistry, Principles, and Commercial Applications; Banks, R. E.; Smart, B. E.; Tatlow, C. J., Eds.; Springer: New York, 1994; pp 237–262. |
| 27. | Brisdon, A. K.; Crossley, I. R.; Pritchard, R. G.; Sadiq, G.; Warren, J. E. Organometallics 2003, 22, 5534–5542. doi:10.1021/om034146t |
| 28. | Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 20, 2420–2421. doi:10.1039/b207979h |
| 14. | Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/C39950000051 |
| 15. | Yamazaki, T.; Mizutani, K.; Kitazume, T. J. Org. Chem. 1995, 60, 6046–6056. doi:10.1021/jo00124a013 |
| 16. | Konno, T.; Chae, J.; Kanda, M.; Nagai, G.; Tamura, K.; Ishihara, T.; Yamanaka, H. Tetrahedron 2003, 59, 7571–7580. doi:10.1016/S0040-4020(03)01199-2 |
| 17. | Chen, Q.; Qiu, X.-L.; Qing, F.-L. J. Fluorine Chem. 2007, 128, 1182–1186. doi:10.1016/j.jfluchem.2007.02.010 |
| 18. | Jiang, Z.-X.; Qin, Y.-Y.; Qing, F.-L. J. Org. Chem. 2003, 68, 7544–7547. doi:10.1021/jo0344384 |
| 19. | Xiong, Z.; Qiu, X.-L.; Huang, Y.; Qing, F.-L. J. Fluorine Chem. 2011, 132, 166–174. doi:10.1016/j.jfluchem.2010.12.012 |
| 20. | Hanamoto, T.; Hakoshima, Y.; Egashira, M. Tetrahedron Lett. 2004, 45, 7573–7576. doi:10.1016/j.tetlet.2004.08.118 |
| 21. | Chen, X.-Y.; Qiu, X.-L.; Qing, F.-L. Tetrahedron 2008, 64, 2301–2306. doi:10.1016/j.tet.2008.01.021 |
| 22. | Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073–4076. doi:10.1021/ol048229x |
| 23. | Watanabe, Y.; Yamazaki, T. J. Org. Chem. 2011, 76, 1957–1960. doi:10.1021/jo102503s |
| 24. | Yamazaki, Y.; Yamamoto, T.; Ichihara, R. J. Org. Chem. 2006, 71, 6251–6253. doi:10.1021/jo060909l |
| 25. | Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945–5948. doi:10.1016/j.tet.2009.05.087 |
| 26. | Konno, T.; Moriyasu, K.; Ishihara, T. Synthesis 2009, 1087–1094. doi:10.1055/s-0028-1087988 |
| 11. | Kobayashi, Y.; Kumadaki, I. Tetrahedron Lett. 1969, 10, 4095–4096. doi:10.1016/S0040-4039(01)88624-X |
| 12. | Oishi, M.; Kondo, H.; Amii, H. Chem. Commun. 2009, 1909–1911. doi:10.1039/b823249k |
| 13. | Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679–1681. doi:10.1126/science.1190524 |
| 7. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 8. | Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A. J. Am. Chem. Soc. 1989, 111, 393–395. doi:10.1021/ja00183a073 |
| 9. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 10. | Ma, J.-A.; Cahard, D. J. Fluorine Chem. 2007, 128, 975–996. doi:10.1016/j.jfluchem.2007.04.026 |
| 33. | Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. Angew. Chem., Int. Ed. 2012, 51, 6467–6470. doi:10.1002/anie.201200827 |
| 34. | Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56–61. doi:10.1016/j.jfluchem.2013.01.004 |
| 31. | Omote, M.; Tanaka, M.; Ikeda, A.; Nomura, S.; Tarui, A.; Sato, K.; Ando, A. Org. Lett. 2012, 14, 2286–2289. doi:10.1021/ol300670n |
| 32. | Omote, M.; Tanaka, M.; Tanaka, M.; Ikeda, A.; Tarui, A.; Sato, K.; Ando, A. J. Org. Chem. 2013, 78, 6196–6201. doi:10.1021/jo400859s |
| 30. | Prakash, G. K. S.; Krishnan, H. S.; Jog, P. V.; Iyer, A. P.; Olah, G. A. Org. Lett. 2012, 14, 1146–1149. doi:10.1021/ol300076y |
| 29. | Yamazaki, T.; Takita, K.; Ishikawa, N. J. Fluorine Chem. 1985, 30, 357–363. doi:10.1016/S0022-1139(00)84971-4 |
| 35. |
Qing, F.; Gao, W. Chin. J. Org. Chem. 2000, 20, 764–768.
http://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract333670.shtml |
© 2013 Ikeda et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)