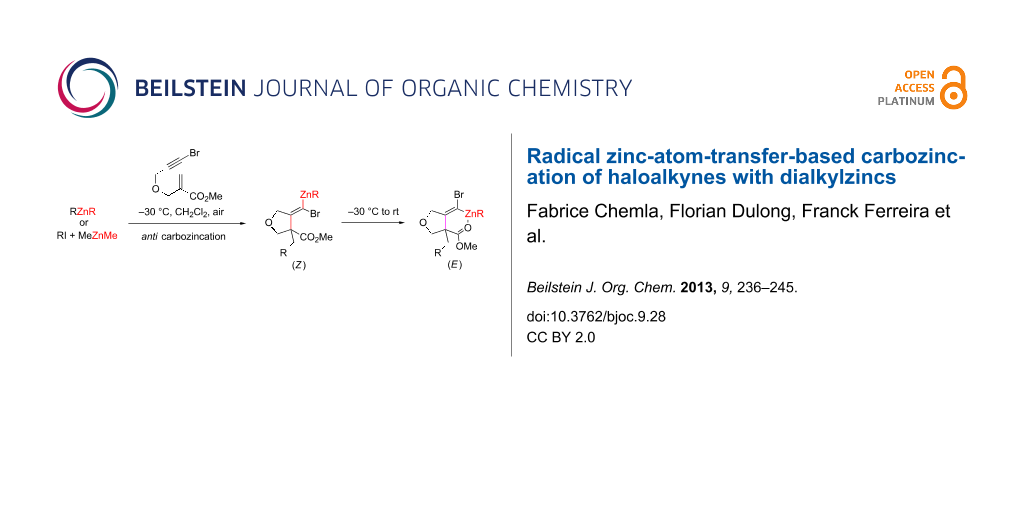
Abstract
The formation of alkylidenezinc carbenoids by 1,4-addition/carbozincation of dialkylzincs or alkyl iodides based on zinc atom radical transfer, in the presence of dimethylzinc with β-(propargyloxy)enoates having pendant iodo- and bromoalkynes, is disclosed. Formation of the carbenoid intermediate is fully stereoselective at −30 °C and arises from a formal anti-selective carbozincation reaction. Upon warming, the zinc carbenoid is stereochemically labile and isomerizes to its more stable form.

Graphical Abstract
Introduction
The last few years have witnessed a gaining interest in the use of organozinc reagents as nontoxic radical precursors or mediators [1-3]. As part of this development, the so-called radical-polar reactions in which alkylzinc reagents are used as mediators in a radical transformation that affords a new zincated species, have emerged as valuable tools in synthesis. Pivotal to the processes disclosed so far using alkylzinc derivatives is zinc atom radical transfer [4]. In general terms, the reaction involves a radical chain process initiated by the formation of an alkyl radical from the organozinc derivative in the presence of oxygen [5-14]. The newly formed radical then undergoes one or more radical transformations before being reduced by the alkylzinc reagent through homolytic substitution at zinc, producing a new organozinc derivative along with an alkyl radical that sustains a radical chain. Overall, the in situ transformation of simple organozinc reagents into more elaborate ones is thus achieved, and subsequent reaction with electrophiles is possible [15-30].
More specifically, building on well-established addition reactions of carbon-centered radicals to carbon–carbon double and triple bonds, such reactivity has been advantageously employed in the context of carbozincation chemistry [31]. The intramolecular carbozincation of unactivated terminal alkenes following zinc atom transfer processes, including a 5-exo-trig cyclization step, has been reported. This is, for instance, the case in the formation of (pyrrolidylmethyl)zinc and (tetrahydrofuranylmethyl)zinc derivatives by reaction of dialkylzinc, organozinc and copper–zinc mixed reagents with (N-allyl)aminoenoates [32-34] and β-(allyloxy)enoates [35], in the formation of (pyrrolidonylmethyl)zinc by condensation of dialkylzincs with N,N-diallylpropiolamide [36], and also in the cyclization of alkenylzinc iodides to cyclopentylmethylzinc iodides, formerly believed to be anionic in nature [4]. Carbozincations of alkynes based on zinc atom transfer have also been disclosed. The reaction of dialkylzincs or of alkyl iodides in the presence of Me2Zn/O2 with β-(propargyloxy)enoates entails the intramolecular carbometallation of the pendant alkynes substituted by silyl, alkyl, aryl, alkenyl or amino groups by a 5-exo dig radical cyclization step [37,38]. Intermolecular carbozincation of terminal arylacetylenes [39] and of diethyl acetylenedicarboxylate [40] has been achieved by dialkylzinc-mediated radical additions. Worthy of note is that in some cases the zinc-atom-transfer-based carbozincation of alkynes can occur with anti selectivity [38,40], and thereby represents a complementary approach to transition-metal-mediated carbozincations, which are generally syn-selective [41-46].
To explore further the possibilities offered by zinc atom transfer processes we considered the possibility to prepare alkylidenezinc carbenoids by radical-based carbozincation of haloalkynes. Such carbenoids are multipurpose reagents [47] that are typically prepared from 1,1'-dihaloalkenes, either by lithium/halogen exchange followed by transmetallation with zinc salts or by direct zinc/halogen exchange [48-50]. Alternatively, they can also be prepared efficiently by selective monohalogenation of alkylidene gem-bismetallic intermediates [51].
To the best of our knowledge, the preparation of alkylidenezinc carbenoids by the direct carbozincation of haloalkynes has not been reported [52]. As a starting point to develop such an approach, we reasoned that the reaction of dialkylzincs with β-(propargyloxy)enoates bearing pendant haloalkynes would be ideally suited (Scheme 1). On the one hand it would provide a means to control totally the regioselectivity of the radical addition to the haloalkyne, and on the other hand the envisioned zinc atom transfer to an α-halo vinylic radical should be favorable as a result of the presence of the ester moiety. Hereafter, we disclose our findings concerning this reaction.
![[1860-5397-9-28-i1]](/bjoc/content/inline/1860-5397-9-28-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Anticipated formation of alkylidene zinc carbenoids by reaction of dialkylzincs with β-(propargyloxy)enoates having pendant haloalkynes.
Scheme 1: Anticipated formation of alkylidene zinc carbenoids by reaction of dialkylzincs with β-(propargylox...
Results and Discussion
β-(Propargyloxy)enoates 3a and 3b having a pendant bromoalkyne and an iodoalkyne moiety, respectively, were prepared by condensation of propargylic alcohols 1 with methyl 2-(bromomethyl)acrylate (2) (Scheme 2). Enoate 3a was readily obtained by direct reaction of 3-bromopropargyl alcohol (1a). By contrast, the reaction of the iodo analogue 1b with 2 proved troublesome as it led to inseparable mixtures of the desired enoate 3b and non-iodinated enoate 3c. Thus, 3b was best prepared by iodinating (AgNO3/NIS) the terminal alkyne of enoate 3c prepared from propargyl alcohol (1c) and acrylate 2.
![[1860-5397-9-28-i2]](/bjoc/content/inline/1860-5397-9-28-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of β-(propargyloxy)enoates having pendant haloalkynes. Reagents and conditions: (a) 2 (1.4 equiv), Et3N (4 equiv), NaI (10 mol %), CH2Cl2, 50 °C, sealed tube, 74% (3a), 87% (3c); (b) AgNO3 (1.5 equiv), N-iodosuccinimide (1.5 equiv), acetone, rt, 53% (3b).
Scheme 2: Preparation of β-(propargyloxy)enoates having pendant haloalkynes. Reagents and conditions: (a) 2 (...
According to our previously optimized conditions for the 1,4-addition/carbozincation reaction of dialkylzincs with β-(propargyloxy)enoates [37,38], bromoalkyne 3a was treated with Et2Zn at room temperature in Et2O under an argon atmosphere (Table 1, entry 1). To our delight, following acidic work-up, the expected methylenetetrahydrofuranyl bromide 4aa was obtained in 43% isolated yield as a mixture of diastereoisomers in a 77:23 Z/E ratio [53]. Hydrolysis with D2O evidenced the intermediate formation of an alkylidene zinc carbenoid as deuterated 4aa-D was produced (Table 1, entry 2). As previously noted in the case of similar 1,4-addition/carbozincation sequences [37,38], deuterium incorporation was nearly quantitative for the Z isomer, and much lower for the E one. More unexpectedly, however, 40% of alkylidenetetrahydrofuran 5a, wherein the bromine atom had been substituted by an ethyl group, was also isolated as a 79:21 Z/E mixture. Deuterium labeled 5a-D was produced following hydrolysis with D2O (Table 1, entry 2), thereby showing that an alkylidenezinc intermediate was being formed in the generation of this side-product under these reaction conditions.
Table 1: 1,4-addition/carbozincation of dialkylzincs with β-(propargyloxy)enoates 3 having pendant haloalkynes in the presence of traces of air.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-28-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Enoate | X | Solvent | R | Products (Yieldb (%) [drc (Z/E)]) |
|---|---|---|---|---|---|
| 1 | 3a | Br | Et2O | Et | 4aa (43 [77:23]); 5a (40 [79:21]) |
| 2 | 3a | Br | Et2O | Et |
4aa-Dd (44 [81(90% D):19(10% D)]);
5a-Dd (35 [84(85% D):16(40% D)]) |
| 3 | 3b | I | Et2O | Et | 4ba (40e [98:2]); 5a (21e [82:18]) |
| 4 | 3a | Br | Et2O | n-Bu | 4ab (76 [70:30]); 5b (7 [ndf]) |
| 5 | 3b | I | Et2O | n-Bu | 4bb (58 [98:2]) |
| 6 | 3a | Br | CH2Cl2 | Et | 4aa (39d [78:22]); 5a (17d [91:9]) |
| 7 | 3a | Br | CH2Cl2 | n-Bu | 4ab (37 [76:24]); 5b (12 [nd]f) |
aReaction conditions : R2Zn (3 equiv), rt, 24 h under Ar atmosphere (see Experimental section).
bCombined yield of diastereomers after chromatography unless otherwise noted.
cDetermined by 1H NMR analysis of the crude material.
dThe reaction mixture was quenched with D2O. The percentage of deuterium incorporation is given in parenthesis for each compound.
eDetermined by 1H NMR spectroscopy based on analysis of the crude mixture with biphenyl as internal standard.
fNot determined.
When iodoalkyne 3b was used, a similar 40% yield of vinylic iodide 4ba was obtained, but this time exclusively as the Z isomer (Table 1, entry 3). 5a was also produced, but in a lower 21% yield and similar diastereoselectivity (82:18 Z/E ratio). Significantly lower levels of side-product formation arising from halogen substitution were observed when n-Bu2Zn was used, thus leading to improved results (Table 1, entries 4 and 5). The reaction with bromoalkyne 3a provided vinylic bromide 4ab in 76% yield and 70:30 Z/E ratio and only 7% of 5b. Better, the reaction with iodoalkyne 3b afforded exclusively iodide 4bb in 58% yield and complete diastereoselectivity in favor of the (Z) isomer. Formation of substitution side-products was also diminished when CH2Cl2 was used as the solvent instead of Et2O, even though this had little impact on the efficiency and diastereoselectivity of vinyl halide formation (Table 1, entries 6 and 7). Reaction of Et2Zn with 3a provided vinyl bromide 4aa in 39% yield (78:22 Z/E ratio) and 5a in 17% yield, while reaction of n-Bu2Zn gave 4ab in 37% yield (76:24 Z/E ratio) and 5b in 12% yield. It is worthy of note that no difference was observed between the different dialkylzincs in this case.
The formation of alkylidenezinc derivatives 7 leading to compounds 5 is intriguing (Scheme 3). A first possible mechanistic route could involve the reaction of zinc carbenoid 6 and the excess of dialkylzinc reagent via the intermediate formation of a zincate [48-51] (Scheme 3, path a). The stereoselectivity of such rearrangements is often dependent on the substrate structure, so the diastereopurity of 5 is not necessarily informative about that of 6 [48-51]. An alternative possibility to account for the formation of 7 could be the reaction of the dialkylzinc reagent with enoate 8 arising from a prior substitution of bromoalkyne 3a with the dialkylzinc reagent (Scheme 3, path b). Both the diastereoselectivity and the levels of deuterium incorporation are very close to those obtained for the reaction of diethylzinc with pure 8 [37], which argues in favor of this mechanistic scenario.
![[1860-5397-9-28-i3]](/bjoc/content/inline/1860-5397-9-28-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible reaction pathways to account for the formation of product 5.
Scheme 3: Possible reaction pathways to account for the formation of product 5.
To try to discriminate between these possibilities we conducted some additional test experiments (Scheme 4). In agreement with the general consideration that dialkylzinc reagents do not undergo uncatalyzed cross-coupling reactions with bromoalkynes, no reaction was observed between Et2Zn and 1-bromohexyne (9) [54]. By contrast, bromoalkyne 11 having a silyloxy group at the propargylic position reacted smoothly to afford ethyl-substituted alkyne 10 along with alkene 12, which had incorporated two ethyl groups. 12 was isolated as a mixture of diastereoisomers in 70:30 dr. The fact that no reaction takes place between pure 10 and Et2Zn indicates that 12 is not formed by carbozincation. Hence, most likely it is formed by the reaction of Et2Zn and carbenoid 13 arising from the carbozincation of 11 (Scheme 4). Moreover, if 13 is indeed formed, it would also lead to alkyne 10 following Fritsch–Buttenberg–Wieschell (FBW) rearrangement [55-57]. Since the presence of the oxygen atom in the propargylic position should facilitate the carbometallation reaction [58], this mechanistic pathway provides a plausible explanation for the fact that bromine substitution occurs from α-oxgenated bromoalkyne 11 and not from 9.
![[1860-5397-9-28-i4]](/bjoc/content/inline/1860-5397-9-28-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Test experiments to gain insight into the mechanism of formation of alkylidene zinc intermediate 7.
Scheme 4: Test experiments to gain insight into the mechanism of formation of alkylidene zinc intermediate 7.
Regarding our 1,4 addition/carbocyclization sequence, these test experiments provide two important pieces of evidence for the behavior of 3a in the presence of a dialkylzinc. First, β-alkoxy bromoalkynes undergo direct substitution with Et2Zn to some extent. Second, alkylidenezinc carbenoids react with dialkylzincs to afford the bromine substitution product. Thus, formation of alkylidenezinc compound 7 (and thereby 5) most probably arises from both depicted mechanistic pathways (paths a and b, Scheme 3). In such a situation, we reasoned that in both possibilities, reducing the reaction time would limit the production of the unwanted side-products by limiting the contact time between the dialkylzinc reagent and either the starting bromoalkyne or the generated zinc carbenoid. Thus, we considered adding air to the reaction media in order to accelerate the oxidation of the dialkylzinc species and therefore radical production (Table 2).
Table 2: 1,4-Addition/carbozincation of dialkylzincs on 3a in the presence of added air.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-28-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R | Reaction conditions | Products [ratio] | Yieldb (%) | drc of product 4 (E/Z) |
|---|---|---|---|---|---|
| 1 | Et | rt, 1 h | 4aa/(Z)-5a [84:16] | 69d | 54:46 |
| 2 | Et | 0 °C, 1 h | 4aa | 93 | 87:13 |
| 3 | Et | −30 °C, 1 h | 4aa | 89 | >98:2 |
| 4 | Et | −30 °C, 1 h | 4aa-De | 95d | >98(83% D):2 |
| 5 | Et | −30 °C, 1 h, DCEf | 4aa-De | 89 | >98(83% D):2 |
| 6 | Bu | −30 °C, 1 h | 4ab | 93 | >98:2 |
| 7 | Et | −30 °C, 1 h then rt, 24h | 4aa/(Z)-5a [78:22] | 78d | 44:56 |
| 8 | Et | −30 °C, 1 h then rt, 24 h | 4aa-De/(Z)-5a-Dg [78:22] | 78d | 44(<10% D):56(<10% D) |
aReaction conditions : R2Zn (3 equiv), CH2Cl2, dry air was bubbled at once into the reaction mixture, which was then kept under Ar atmosphere (see Experimental section).
bCombined yield of products after chromatography unless otherwise noted.
cDetermined by 1H NMR analysis of the crude material.
dDetermined by 1H NMR spectroscopy based on analysis of the crude mixture with biphenyl as the internal standard.
eThe reaction mixture was quenched with D2O. The percentage of deuterium incorporation is given in parenthesis for each isomer.
fDCE = 1,2-dichloroethane was used as solvent instead of CH2Cl2.
g30% deuterium incorporation was observed for product 5a-D.
A reduced amount of side-product 5 was indeed observed in the reaction of enoate 3a with Et2Zn in CH2Cl2 at room temperature in the presence of added dry air (Table 2, entry 1). A mixture of vinyl bromide 4aa and alkene 5a in a 84:16 ratio and in 69% overall yield was obtained. However the diastereoselectivity of the formation of 4aa dropped significantly. Lowering the reaction temperature had a highly beneficial impact on the reaction outcome. At 0 °C, the formation of 5a was totally suppressed, and 4aa was obtained with a much better diastereoselectivity, though remarkably in favor of the E isomer (Table 2, entry 2). At −30 °C, the exclusive and totally diastereoselective formation of (E)-4aa in excellent 89% isolated yield was obtained (Table 2, entry 3). Hydrolysis with D2O led to (E)-4aa-D with 83% deuterium incorporation when either CH2Cl2 or DCE were used as solvent (entries 4 and 5), therefore evidencing the intermediate stereoselective formation of an alkylidene zinc carbenoid. Similar results were obtained by using n-Bu2Zn, indicating that the process is quite general (Table 2, entry 6).
The diastereoselectivity of the formation of 4aa seemed to be dependent not only on the reaction temperature, but also on the total reaction time (compare Table 1, entry 6 and Table 2, entry 1). Suspecting a possible Z/E isomerization of the alkylidenezinc carbenoid intermediate, we conducted an experiment wherein air was added to a mixture of enoate 3a and Et2Zn in CH2Cl2 at −30 °C, and the reaction was first kept for 1 h at this temperature and then for 23 h at room temperature (Table 2, entry 7). Following acidic quench, very similar results to those noted for the same reaction carried out at room temperature (Table 2, entry 1) were observed. 4aa was recovered in 61% yield as a 56:44 mixture of isomers along with alkene (Z)-5a in 22% yield. It is worthy to note that very low levels of deuterium incorporation were observed in this case.
These results have a three-fold consequence. First, they indicate that Z/E isomerization of the alkylidenezinc carbenoid occurs between −30 °C and room temperature. Second, it demonstrates that alkene 5a can be formed by the reaction between the zinc carbenoid and Et2Zn (Scheme 3, path a), and that in such a case the transformation is stereoselective. Third, when oxygen is introduced into the reaction media, the alkylidenezinc carbenoid is eventually demetallated upon standing at room temperature.
The different results obtained for the 1,4-addition/carbozincation of enoates having pendant bromoalkynes are consistent with our anticipated zinc atom radical transfer mechanism (Scheme 1) and can be rationalized according to the following scenario (Scheme 5). The process involves the initial 1,4-addition of radical R to the starting enoate and the subsequent 5-exo-dig cyclisation of enoxy radical 14 to provide α-bromovinyl radical 15 of E geometry. Substitution by electron-withdrawing groups slows down E to Z isomerization of vinylic radicals, and therefore, due to the presence of the bromine atom, interconversion of (E)-15 into (Z)-15 should not be fast. Thus, (E)-15 reacts by Zn atom transfer prior to its equilibration to provide stereoselectively carbenoid (Z)-6 [59] and to some extent by H-atom transfer to give reduced bromoalkene (E)-4 [60]. Upon warming, (Z)-6 isomerizes to its more stable isomer (E)-6 wherein the zinc atom is coordinated intramolecularly to the adjacent ester. To a minor extent, reaction with the excess dialkylzinc present in the reaction media provides alkylidenezinc (E)-7 stereoselectively. Note, however, that in the case where reactions are conducted at room temperature, E/Z equilibration of the intermediate radical 15 should be faster and zinc atom transfer from (Z)-15 may also contribute to the formation of (E)-6.
![[1860-5397-9-28-i5]](/bjoc/content/inline/1860-5397-9-28-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanistic rationale for the reaction of dialkylzincs with β-(propargyloxy)enoate 3a.
Scheme 5: Mechanistic rationale for the reaction of dialkylzincs with β-(propargyloxy)enoate 3a.
Two situations are next to be distinguished. Under the conditions involving excess air, carbenoid 6 is protonated in the reaction media (even though we have not identified the proton source). It is possible that protonation occurs prior to full E to Z isomerization and vinyl bromide 4 is obtained in low diastereoselectivity. Under the conditions involving only a trace of air, after 24 h at room temperature, carbenoid 6 is still present and total Z to E isomerization has occurred. The reason why vinyl bromide 4 is only isolated with moderate diastereoselectivity following hydrolysis, is that (E)-4, formed from (E)-15 by H-abstraction, is present from the start.
We finally considered the prospect to carry out the carbozincation of bromoalkynes using a combination of an alkyl iodide and dimethylzinc (Table 3). Towards this end, we first treated enoate 3a with Me2Zn, following our previously reported optimized conditions for the 1,4-addition/carbozincation of β-(propargyloxy)enoates with Me2Zn [38], which proceeds in CH2Cl2 at 0 °C and under continuous introduction of dry air over a period of 1 h (Table 3, entry 1). After acidic quench, vinylic bromide (E)-4ac [61] was isolated in 77% yield as a single diastereoisomer. The stereoselective formation of an alkylidenezinc carbenoid intermediate was this time evidenced by treating the reaction mixture with iodine (Table 3, entry 2). Vinylic dihalide 16 was isolated in 64% yield, again as a single Z diastereoisomer. Thus, Me2Zn showed a similar reactivity to its higher homologues Et2Zn and n-Bu2Zn, with the additional advantage that no direct formation of vinyl bromide (E)-4ac by hydrogen abstraction was observed.
Table 3: Me2Zn-mediated 1,4-addition/carbozincation of alkyl iodides with 3a in the presence of added air.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-28-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | iPrI (equiv) | E-X | Yieldb (%) | Products [ratio] |
|---|---|---|---|---|
| 1 | 0 | H2O | 77 | 4acc |
| 2 | 0 | I2 | 64 | 16 |
| 3d | 5 | H2O | 59 | 4ad/4ac [87:13] |
| 4d | 10 | H2O | 87 | 4ad/4ac/17 [75:6:19] |
| 5d | 10 | D2O | 75 | 4ad-D/4ac-D/17 [75 (95% D):6 (95% D):19] |
aReaction conditions: Me2Zn (5 equiv), iPrI (equiv), CH2Cl2, 0 °C, 20 mL dry air was bubbled during 1 h into the reaction mixture via a syringe pump.
bCombined yield of isolated products after chromatography unless otherwise noted.
cThe product was contaminated with ~10% of product resulting from the addition of the dichloromethyl radical (4ae, R = CHCl2).
d3 equiv Me2Zn were used.
In the presence of 5 equiv iPrI, a mixture of two (diastereomerically pure) compounds 4ad and 4ac in 87:13 ratio was observed. Incorporation of the iPr moiety was therefore the major reaction pathway. The competitive addition of a Me group could be reduced by increasing the amount of iPrI to 10 equiv (Table 3, entries 3 and 4). However, in this case, significant amounts of vinylic dihalide 17 were also isolated. Thus, if large amounts of iodide are used, radical iodine atom transfer between iPrI and the α-bromovinylic radical 15 resulting from the cyclization step (Scheme 5) becomes competitive with the desired zinc atom transfer and hampers the carbozincation process.
Conclusion
In conclusion, we have shown that β-(propargyloxy)enoates having pendant iodo- and bromoalkynes undergo a 1,4-addition/carbozincation sequence by reaction with dialkylzincs or with alkyliodides in the presence of dimethylzinc. The sequence involves a radical chain mechanism initiated by air and provides the proof of concept that alkylidenezinc carbenoids can be prepared by carbozincation based on zinc atom transfer. In the disclosed process, we have demonstrated that the formation of a bromocarbenoid intermediate is fully stereoselective at −30 °C and arises from a formal anti-selective carbozincation reaction. Upon warming, the zinc carbenoid is stereochemically labile and isomerizes to its more stable form. In the absence of added air, no decomposition of the carbenoid intermediate is observed at room temperature for at least 24 h. Deuterium labeling and iodolysis experiments evidence that the zinc carbenoids prepared under such reactions can act as typical nucleophiles and should, thus, be well-suited for subsequent functionalization [47-51]. Furthermore, as indicated with the formation of some side-products observed during this work, they should also react readily as electrophiles toward organometallic nucleophiles and undergo intramolecular nucleophilic substitution reactions [47-51].
Experimental
Experiments involving organometallic compounds were carried out in dried glassware under a positive pressure of dry Ar. All solvents were distilled to remove stabilizers and dried with a MBRAUN Solvent Purification System SPS-800. n-Bu2Zn (Fluka, ~1 N in heptane), Et2Zn (Aldrich, 1.0 M in hexanes), Me2Zn (Aldrich, 1.0 M in heptane) and all other reagents were of commercial quality and were used without purification. 1H NMR, 13C NMR spectra were recorded with a Bruker AVANCE 400 spectrometer fitted with BBFO probe with Z gradient. Chemical shifts are reported in δ relative to an internal standard of residual chloroform (δ 7.27 for 1H NMR and 77.16 for 13C NMR). IR spectra were recorded with an ATR diamond spectrophotometer. High-resolution mass spectra (HRMS) were obtained on a Finnigan MAT 95.
General Procedure 1. Reaction of n-Bu2Zn and Et2Zn with β-(propargyloxy)enoates 3a and 3b having pendant haloalkynes in the presence of a trace amount of air (Table 1): Under argon, to a stirred solution of β-(propargyloxy)enoate (0.2 mmol) in Et2O (1 mL) at room temperature was added R2Zn (0.6 mmol). The reaction mixture was stirred at room temperature for 24 h. The reaction was hydrolyzed with an aqueous solution of HCl (1 M, 10 mL). The layers were separated, the aqueous one being extracted with Et2O (2 × 15 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure, and the residue was purified by flash chromatography (pentane/ether).
General Procedure 2. Reaction of n-Bu2Zn and Et2Zn with β-(propargyloxy)enoate 3a in the presence of added air (Table 2): Under argon, to a stirred solution of β-(propargyloxy)enoate 3a (0.2 mmol) in CH2Cl2 (2 mL) at −30 °C was added R2Zn (0.6 mmol). Air (2 mL) was bubbled at once into the solution via a syringe fitted with a CaCl2 guard and the reaction mixture was stirred at −30 °C for 1 h. CH2Cl2 (20 mL) and an aqueous solution of HCl (1 M, 10 mL) were added to quench the reaction. The layers were separated, the aqueous one being extracted with Et2O (2 × 15 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure to afford the crude product.
(Z)-Methyl 4-(bromomethylene)-3-propyltetrahydrofuran-3-carboxylate ((Z)-4aa): Prepared according to general procedure 1 from enoate 3a (50 mg, 0.2 mmol) and Et2Zn (0.6 mL, 1.0 M in hexanes, 0.6 mmol). Purification by flash chromatography with pentane/ether (95:05) as eluent gave the title compound ((Z)-4aa) (19 mg, 33%) as a colorless oil. IR (neat): 2958, 1730, 1434, 1219, 1072, 938, 722 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.34 (t, J = 2.7 Hz, 1H), 4.42 (d(AB system), J = 9.1 Hz, 1H), 4.40 (d br, J = 2.9 Hz, 2H), 3.84 (d(AB system), J = 9.1 Hz, 1H), 3.74 (s, 3H), 1.90 (m, 1H), 1.68 (m, 1H), 1.24 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.3, 146.6, 100.4, 75.7, 72.8, 58.5, 52.6, 39.5, 18.8, 14.2; HRMS–ESI (m/z): [M + Na]+ calcd for C10H15O3BrNa: 285.00968; found: 285.00990.
(E)-Methyl 4-(bromomethylene)-3-propyltetrahydrofuran-3-carboxylate ((E)-4aa): Prepared according to general procedure 2 from enoate 3a (47 mg, 0.2 mmol) and Et2Zn (0.6 mL, 1.0 M in hexanes, 0.6 mmol). The title compound ((E)-4aa) was isolated pure (47 mg, 89%) as a colorless oil and did not require further purification. IR (neat): 2960, 2873, 1769, 1730, 1433, 1217, 1122, 1074, 968, 938, 742 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.15 (t, J = 1.9 Hz, 1H), 4.39 (dd(ABX system), J = 12.7, 1.9 Hz, 1H), 4.32 (dd(ABX system), J = 12.7, 1.8 Hz, 1H), 4.12 (d(AB system), J = 8.9 Hz, 1H), 3.97 (d(AB system), J = 8.9 Hz, 1H), 3.74 (s, 3H), 2.13 (td, J = 13.5, 4.6 Hz, 1H), 1.90 (dt, J = 12.5, 4.4 Hz, 1H), 1.50–1.30 (m, 1H), 1.30–1.25 (m, 1H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.3, 146.7, 98.5, 78.3, 73.8, 58.3, 52.7, 34.7, 17.9, 14.8; HRMS–ESI (m/z): [M + Na]+ calcd for C10H15O3BrNa: 285.0097; found: 285.0091.
(Z)-Methyl 4-(iodomethylene)-3-pentyltetrahydrofuran-3-carboxylate ((Z)-4bb): Prepared according to general procedure 1 from enoate 3b (40 mg, 0.14 mmol) and Bu2Zn (0.42 mL, ~1 N in heptane, 0.42 mmol). Purification by flash chromatography with pentane/ether (80:20) as eluent gave the title compound ((Z)-4bb) (28 mg, 58%) as a colorless oil. IR (neat): 2951, 2925, 2856, 1731, 1434, 1230, 1073, 939, 730 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.36 (t, J = 2.6 Hz, 1H), 4.49 (d(AB system), J = 9.1 Hz, 1H), 4.31 (d br, J = 2.6 Hz, 2H), 3.90 (d(AB system), J = 9.1 Hz, 1H), 3.74 (s, 3H), 1.90 (m, 1H), 1.66 (m, 1H), 1.40–1.20 (m, 6H), 0.91 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.3, 152.4, 76.5, 76.2, 71.3, 59.7, 52.7, 37.3, 32.0, 25.1, 22.5, 14.1.
(E)-Methyl 4-(bromomethylene)-3-pentyltetrahydrofuran-3-carboxylate ((E)-4ab): Prepared according to general procedure 2 from enoate 3a (47 mg, 0.2 mmol) and Bu2Zn (0.6 mL, ~1 N in heptane, 0.6 mmol). The title compound ((E)-4ab) was isolated pure (55 mg, 93%) as a colorless oil and did not require further purification. IR (neat): 2955, 2927, 2856, 1771, 1733, 1433, 1260, 1230, 1217, 1122, 1074, 936, 910, 731 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.15 (t, J = 1.9 Hz, 1H), 4.39 (dd(ABX system), J = 12.7, 2.0 Hz, 1H), 4.32 (dd(ABX system), J = 12.7, 1.8 Hz, 1H), 4.12 (d(AB system), J = 8.9 Hz, 1H), 3.97 (d(AB system), J = 8.9 Hz, 1H), 3.74 (s, 3H), 2.15 (td, J = 13.7, 4.2 Hz, 1H), 1.91 (dt, J = 12.2, 4.4 Hz, 1H), 1.50–1.20 (m, 6H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.3, 146.7, 98.6, 78.3, 73.9, 58.2, 52.7, 32.5, 32.4, 24.1, 22.7, 14.3; HRMS–ESI (m/z): [M + Na]+ calcd for C12H19O3BrNa: 313.0410; found: 313.0413.
(E)-Methyl 4-(bromomethylene)-3-ethyltetrahydrofuran-3-carboxylate ((E)-4ac) (Table 3, entry 1): Under argon, to a stirred solution of β-(propargyloxy)enoate 3a (46 mg, 0.2 mmol) in CH2Cl2 (1 mL) at 0 °C was added Me2Zn (1 mL, 1.0 M in heptane, 1.0 mmol). Air (20 mL) was slowly introduced over 1 h into the solution via a syringe pump by using a syringe fitted with a CaCl2 guard. The reaction mixture was then stirred at 0 °C for 1 h. CH2Cl2 (5 mL) was then added, and the reaction was hydrolyzed with an aqueous solution of HCl (1 M, 5 mL). The layers were separated, the aqueous one being extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography on silica gel (pentane/ether 80:20) afforded the title compound ((E)-4ac) (38 mg, 77%) as a colorless oil. IR (neat): 2953, 1767, 1731, 1638, 1229, 1138, 1034, 936, 786 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.17 (t, J = 1.9 Hz, 1H), 4.41 (dd(ABX system), J = 12.7, 1.9 Hz, 1H), 4.33 (dd(ABX system), J = 12.7, 1.9 Hz, 1H), 4.13 (d(AB system), J = 8.9 Hz, 1H), 3.98 (d(AB system), J = 8.9 Hz, 1H), 3.75 (s, 3H), 2.21 (m, 1H), 2.00 (m, 1H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.2, 146.4, 98.5, 77.9, 73.8, 58.5, 52.6, 25.2, 8.9; HRMS–ESI (m/z): [M + Na]+ calcd for C9H13O3BrNa: 270.99403; found: 270.99477.
(E)-Methyl 4-(bromomethylene)-3-isobutyltetrahydrofuran-3-carboxylate ((E)-4ad) (Table 3, entry 4): Under argon, to a stirred solution of β-(propargyloxy)enoate 3a (47 mg, 0.2 mmol) and iPrI (0.2 mL, 2.0 mmol) in CH2Cl2 (1 mL) at 0 °C was added Me2Zn (0.6 mL, 1.0 M in heptane, 0.6 mmol). Air (20 mL) was slowly introduced over 1 h into the solution via a syringe pump by using a syringe fitted with a CaCl2 guard. The reaction mixture was then stirred at 0 °C for 1 h. CH2Cl2 (5 mL) was then added and the reaction was hydrolyzed with an aqueous solution of HCl (1 M, 5 mL). The layers were separated, the aqueous one being extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography on silica gel (pentane/ether 50:50) afforded the title compound ((E)-4ad) (36 mg, 65%) as a colorless oil. IR (neat): 2953, 2870, 1732, 1640, 1230, 1126, 1073, 937, 695 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.15 (t, J = 1.9 Hz, 1H), 4.40 (d, J = 1.9 Hz, 2H), 4.15 (d(AB system), J = 8.9 Hz, 1H), 4.02 (d(AB system), J = 8.9 Hz, 1H), 3.74 (s, 3H), 2.05 (m, 2H), 1.77 (m, 1H), 0.96 (d, J = 8.4 Hz, 3H), 0.95 (d, J = 8.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.1, 146.9, 98.6, 78.0, 73.6, 58.1, 52.5, 40.5, 24.9, 24.5, 24.4; HRMS–ESI (m/z): [M + Na]+ calcd for C11H17O3BrNa: 299.02533; found: 299.02606.
(Z)-Methyl 4-(bromoiodomethylene)-3-ethyltetrahydrofuran-3-carboxylate (16) (Table 3, entry 2): Under argon, to a stirred solution of β-(propargyloxy)enoate 3a (45 mg, 0.2 mmol) in CH2Cl2 (1 mL) at 0 °C was added Me2Zn (1 mL, 1.0 M in heptane, 1.0 mmol). Air (20 mL) was slowly introduced over 1 h into the solution via a syringe pump by using a syringe fitted with a CaCl2 guard. The reaction mixture was then stirred at 0 °C for 1 h. A solution of I2 (330 mg, 1.3 mmol) in THF (1 mL) was then added at the same temperature, and the mixture was stirred for 1 h. CH2Cl2 (10 mL) followed by an aqueous solution of Na2S2O3 (10%) were added. The layers were separated, the aqueous one being extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed with HCl (1 M) (10 mL) and brine (10 mL), dried over MgSO4, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (pentane/ether 80:20) afforded the title compound 16 (46 mg, 64%) as a pale yellow oil. IR (neat): 2947, 2878, 1730, 1630, 1432, 1236, 1081, 941 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.32 (d(AB system), J = 14.0 Hz, 1H), 4.27 (d(AB system), J = 14.0 Hz, 1H), 4.24 (d(AB system), J = 8.8 Hz, 1H), 4.15 (d(AB system), J = 8.8 Hz, 1H), 3.75 (s, 3H), 2.21 (m, 1H), 1.95 (m, 1H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.4, 152.4, 80.1, 79.5, 60.9, 52.8, 41.2, 25.5, 9.0; HRMS–ESI (m/z): [M + Na]+ calcd for C9H12O3BrINa: 396.89067; found: 396.89159.
(Z)-Methyl 4-(bromoiodomethylene)-3-isobutyltetrahydrofuran-3-carboxylate (17) (Table 3, entry 4): The title compound was obtained as a side-product following the above-described procedure for the preparation of (E)-4ad. It could not be isolated pure after column chromatography, but characteristic NMR spectroscopic data were obtained. 1H NMR (400 MHz, CDCl3) δ 4.32 (d(AB system), J = 14.0 Hz, 1H), 4.29 (d(AB system), J = 14.0 Hz, 1H), 4.24 (d(AB system), J = 8.9 Hz, 1H), 4.21 (d(AB system), J = 8.9 Hz, 1H), 3.75 (s, 3H), 2.10 (dd, J = 14.5, 6.8 Hz, 1H), 1.95 (dd, J = 14.5, 5.0 Hz, 1H), 1.81 (m, 1H), 0.98 (d, J = 6.7 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.5, 152.9, 80.3, 79.8, 58.3, 52.7, 41.9, 40.7, 24.7, 24.5, 23.8.
References
-
Bazin, S.; Feray, L.; Bertrand, M. P. Chimia 2006, 60, 260–265.
Return to citation in text: [1] -
Akindele, T.; Yamada, K.-i.; Tomioka, K. Acc. Chem. Res. 2009, 42, 345–355. doi:10.1021/ar800166q
Return to citation in text: [1] -
Seyferth, D. Organometallics 2001, 20, 2940–2955. doi:10.1021/om010439f
Return to citation in text: [1] -
Cohen, T.; Gibney, H.; Ivanov, R.; Yeh, E. A.-H.; Marek, I.; Curran, D. P. J. Am. Chem. Soc. 2007, 129, 15405–15409. doi:10.1021/ja076554k
Return to citation in text: [1] [2] -
Lewiński, J.; Marciniac, W.; Lipkowski, J.; Justyniak, I. J. Am. Chem. Soc. 2003, 125, 12698–12699. doi:10.1021/ja036020t
Return to citation in text: [1] -
Lewiński, J.; Śliwiński, W.; Dranka, M.; Justyniak, I.; Lipkowski, J. Angew. Chem., Int. Ed. 2006, 45, 4826–4829. doi:10.1002/anie.200601001
Return to citation in text: [1] -
Lewiński, J.; Suwała, K.; Kubisiak, M.; Ochal, Z.; Justyniak, I.; Lipkowski, J. Angew. Chem., Int. Ed. 2008, 47, 7888–7889. doi:10.1002/anie.200803254
Return to citation in text: [1] -
Lewiński, J.; Bury, W.; Dutkiewicz, M.; Maurin, M.; Justyniak, I.; Lipkowski, J. Angew. Chem., Int. Ed. 2008, 47, 573–576. doi:10.1002/anie.200703125
Return to citation in text: [1] -
Lewiński, J.; Kościelski, M.; Suwała, K.; Justyniak, I. Angew. Chem., Int. Ed. 2009, 48, 7017–7020. doi:10.1002/anie.200902716
Return to citation in text: [1] -
Lewiński, J.; Suwała, K.; Kaczorowski, T.; Gałęzowski, M.; Gryko, D. T.; Justyniak, I.; Lipkowski, J. Chem. Commun. 2009, 215–217. doi:10.1039/b813315h
Return to citation in text: [1] -
Jana, S.; Berger, R. J. F.; Fröhlich, R.; Pape, T.; Mitzel, N. W. Inorg. Chem. 2007, 46, 4293–4297. doi:10.1021/ic062438r
Return to citation in text: [1] -
Mileo, E.; Benfatti, F.; Cozzi, P. G.; Lucarini, M. Chem. Commun. 2009, 469–470. doi:10.1039/b818437b
Return to citation in text: [1] -
Maury, J.; Feray, L.; Bazin, S.; Clément, J.-L.; Marque, S. R. A.; Siri, D.; Bertrand, M. P. Chem.–Eur. J. 2011, 17, 1586–1595. doi:10.1002/chem.201002616
Return to citation in text: [1] -
Mukherjee, D.; Ellern, A.; Sadow, A. D. J. Am. Chem. Soc. 2012, 134, 13018–13026. doi:10.1021/ja303440n
Return to citation in text: [1] -
Bertrand, M. P.; Feray, L.; Nouguier, R.; Perfetti, P. J. Org. Chem. 1999, 64, 9189–9193. doi:10.1021/jo9912404
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Vanthuyne, N.; Bertrand, M. P. Tetrahedron 2005, 61, 4261–4274. doi:10.1016/j.tet.2005.02.042
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Vanthuyne, N.; Siri, D.; Bertrand, M. P. Tetrahedron 2007, 63, 77–85. doi:10.1016/j.tet.2006.10.049
Return to citation in text: [1] -
Maury, J.; Feray, L.; Perfetti, P.; Bertrand, M. P. Org. Lett. 2010, 12, 3590–3593. doi:10.1021/ol101519w
Return to citation in text: [1] -
Maury, J.; Mouysset, D.; Feray, L.; Marque, S. R. A.; Siri, D.; Bertrand, M. P. Chem.–Eur. J. 2012, 18, 3241–3247. doi:10.1002/chem.201102366
Return to citation in text: [1] -
Van der Deen, H.; Kellog, R. M.; Feringa, B. L. Org. Lett. 2000, 2, 1593–1595. doi:10.1021/ol005843+
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Yoshida, K.; Takemoto, Y. Synlett 2004, 540–542. doi:10.1055/s-2004-815407
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron 2005, 61, 385–393. doi:10.1016/j.tet.2004.10.104
Return to citation in text: [1] -
Yamada, K.-i.; Umeki, M.; Maekawa, M.; Yamamoto, Y.; Akindele, T.; Nakano, M.; Tomioka, K. Tetrahedron 2008, 64, 7258–7265. doi:10.1016/j.tet.2008.05.069
Return to citation in text: [1] -
Yamada, K.-i.; Konishi, T.; Nakano, M.; Fujii, S.; Cadou, R.; Yamamoto, Y.; Tomioka, K. J. Org. Chem. 2012, 77, 5775–5780. doi:10.1021/jo300944f
Return to citation in text: [1] -
Cozzi, P. G. Angew. Chem., Int. Ed. 2006, 45, 2951–2954. doi:10.1002/anie.200504239
Return to citation in text: [1] -
Cozzi, P. G. Adv. Synth. Catal. 2006, 348, 2075–2079. doi:10.1002/adsc.200606178
Return to citation in text: [1] -
Cozzi, P. G.; Mignogna, A.; Vicennati, P. Adv. Synth. Catal. 2008, 350, 975–978. doi:10.1002/adsc.200700572
Return to citation in text: [1] -
Fernández-Ibáñez, M. A.; Maciá, B.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 1317–1319. doi:10.1002/anie.200704841
Return to citation in text: [1] -
Cozzi, P. G.; Benfatti, F.; Guiteras Capdevila, M.; Mignogna, A. Chem. Commun. 2008, 3317–3318. doi:10.1039/b805197f
Return to citation in text: [1] -
Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. New J. Chem. 2008, 32, 594–606. doi:10.1039/b716292h
Return to citation in text: [1] -
Denes, F.; Chemla, F.; Normant, J. F. Angew. Chem., Int. Ed. 2003, 42, 4043–4046. doi:10.1002/anie.200250474
Return to citation in text: [1] -
Denes, F.; Cutri, S.; Pérez-Luna, A.; Chemla, F. Chem.–Eur. J. 2006, 12, 6506–6513. doi:10.1002/chem.200600334
Return to citation in text: [1] -
Denes, F.; Pérez-Luna, A.; Chemla, F. J. Org. Chem. 2007, 72, 398–406. doi:10.1021/jo061603h
Return to citation in text: [1] -
Giboulot, S.; Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Tetrahedron Lett. 2008, 49, 3963–3966. doi:10.1016/j.tetlet.2008.04.108
Return to citation in text: [1] -
Feray, L.; Bertrand, M. P. Eur. J. Org. Chem. 2008, 3164–3170. doi:10.1002/ejoc.200800242
Return to citation in text: [1] -
Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Chem.–Eur. J. 2008, 14, 8784–8788. doi:10.1002/chem.200801451
Return to citation in text: [1] [2] [3] [4] -
Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993
Return to citation in text: [1] [2] [3] [4] [5] -
Chen, Z.; Zhang, Y.-X.; An, Y.; Song, X.-L.; Wang, Y.-H.; Zhu, L.-L.; Guo, L. Eur. J. Org. Chem. 2009, 5146–5152. doi:10.1002/ejoc.200900858
Return to citation in text: [1] -
Maury, J.; Feray, L.; Bertrand, M. P. Org. Lett. 2011, 13, 1884–1887. doi:10.1021/ol200419x
Return to citation in text: [1] [2] -
Stüdemann, T.; Knochel, P. Angew. Chem., Int. Ed. Engl. 1997, 36, 93–95. doi:10.1002/anie.199700931
Return to citation in text: [1] -
Stüdemann, T.; Ibrahim-Ouali, M.; Knochel, P. Tetrahedron 1998, 54, 1299–1316. doi:10.1016/S0040-4020(97)10226-5
Return to citation in text: [1] -
Gourdet, B.; Lam, H. W. J. Am. Chem. Soc. 2009, 131, 3802–3803. doi:10.1021/ja900946h
Return to citation in text: [1] -
Normant, J. F.; Alexakis, A. Synthesis 1981, 841–870. doi:10.1055/s-1981-29622
Return to citation in text: [1] -
Chinkov, N.; Tene, D.; Marek, I. In Metal-Catalyzed Cross Coupling Reactions, 2nd ed.; Diederich, D.; de Meijere, A., Eds.; Wiley-VCH: New York, 2004; p 395.
Return to citation in text: [1] -
Denès, F.; Pérez-Luna, A.; Chemla, F. Chem. Rev. 2010, 110, 2366–2447. doi:10.1021/cr800420x
Return to citation in text: [1] -
Marek, I. Chem. Rev. 2000, 100, 2887–2900. doi:10.1021/cr990288e
Return to citation in text: [1] [2] [3] -
Harada, T.; Katsuhira, T.; Oku, A. J. Org. Chem. 1992, 57, 5805–5807. doi:10.1021/jo00048a002
Return to citation in text: [1] [2] [3] [4] [5] -
Harada, T.; Katsuhira, T.; Hara, D.; Kotani, Y.; Maejima, K.; Kaji, R.; Oku, A. J. Org. Chem. 1993, 58, 4897–4907. doi:10.1021/jo00070a027
Return to citation in text: [1] [2] [3] [4] [5] -
Harada, T.; Katsuhira, T.; Hattori, K.; Oku, A. Tetrahedron 1994, 50, 7987–8002. doi:10.1016/S0040-4020(01)85284-4
Return to citation in text: [1] [2] [3] [4] [5] -
Creton, I.; Marek, I.; Normant, J.-F. Synthesis 1996, 1499–1508. doi:10.1055/s-1996-4422
Return to citation in text: [1] [2] [3] [4] [5] -
Deng, C.-L.; Song, R.-J.; Liu, Y.-L.; Li, J.-H. Adv. Synth. Catal. 2009, 351, 3096–3100. doi:10.1002/adsc.200900588
The ZnCl2 catalyzed Conia–ene reaction of iodoalkynes that in principle involves a vinylidene zinc carbenoid intermediate has been reported here.
Return to citation in text: [1] -
The Z configuration for the major isomer was determined by NOE experiments on isolated pure (Z)-4aa. An NOE signal (3%) was observed between the vinylic proton and the protons of the methylene group of the propyl chain α to the ester.
Return to citation in text: [1] -
Yeh, M. C. P.; Knochel, P. Tetrahedron Lett. 1989, 30, 4799–4802. doi:10.1016/S0040-4039(01)80511-6
Return to citation in text: [1] -
Creton, I.; Rezaei, H.; Marek, I.; Normant, J. F. Tetrahedron Lett. 1999, 40, 1899–1902. doi:10.1016/S0040-4039(99)00090-8
Return to citation in text: [1] -
Rezaei, H.; Marek, I.; Normant, J. F. Tetrahedron 2001, 57, 2477–2483. doi:10.1016/S0040-4020(01)00069-2
Return to citation in text: [1] -
Rezaei, H.; Yamanoi, S.; Chemla, F.; Normant, J. F. Org. Lett. 2000, 2, 419–421. doi:10.1021/ol991117z
Return to citation in text: [1] -
Fallis, A. G.; Forgione, P. Tetrahedron 2001, 57, 5899–5913. doi:10.1016/S0040-4020(01)00422-7
Return to citation in text: [1] -
Takahashi, K.; Honda, T. Org. Lett. 2010, 12, 3026–3029. doi:10.1021/ol101034s
Zn-atom radical transfer is facilitated by coordination to a Lewis base. In the present case, the bromine atom may be coordinated to the dialkylzinc reagent and thereby activates it towards homolytic substitution. For a similar proposed coordination involving SmI2, see this reference.
Return to citation in text: [1] -
Even though the H-donor has not been identified, competitive H-atom transfer is a frequent side-reaction in the 1,4-addition/cyclization reaction of dialkylzincs with β-(propargyloxy)enoates, see references [37] and [38].
Return to citation in text: [1] -
The stereochemical assignment was done on the basis of the comparison of the NMR data of 4ac and 4aa. Among others, the chemical shift (δ = 6.17 ppm) and coupling constant of the vinylic proton (J = 1.9 Hz) are indicative of the E-configuration.
Return to citation in text: [1]
| 59. |
Takahashi, K.; Honda, T. Org. Lett. 2010, 12, 3026–3029. doi:10.1021/ol101034s
Zn-atom radical transfer is facilitated by coordination to a Lewis base. In the present case, the bromine atom may be coordinated to the dialkylzinc reagent and thereby activates it towards homolytic substitution. For a similar proposed coordination involving SmI2, see this reference. |
| 60. | Even though the H-donor has not been identified, competitive H-atom transfer is a frequent side-reaction in the 1,4-addition/cyclization reaction of dialkylzincs with β-(propargyloxy)enoates, see references [37] and [38]. |
| 38. | Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993 |
| 1. | Bazin, S.; Feray, L.; Bertrand, M. P. Chimia 2006, 60, 260–265. |
| 2. | Akindele, T.; Yamada, K.-i.; Tomioka, K. Acc. Chem. Res. 2009, 42, 345–355. doi:10.1021/ar800166q |
| 3. | Seyferth, D. Organometallics 2001, 20, 2940–2955. doi:10.1021/om010439f |
| 31. | Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. New J. Chem. 2008, 32, 594–606. doi:10.1039/b716292h |
| 15. | Bertrand, M. P.; Feray, L.; Nouguier, R.; Perfetti, P. J. Org. Chem. 1999, 64, 9189–9193. doi:10.1021/jo9912404 |
| 16. | Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e |
| 17. | Bazin, S.; Feray, L.; Vanthuyne, N.; Bertrand, M. P. Tetrahedron 2005, 61, 4261–4274. doi:10.1016/j.tet.2005.02.042 |
| 18. | Bazin, S.; Feray, L.; Vanthuyne, N.; Siri, D.; Bertrand, M. P. Tetrahedron 2007, 63, 77–85. doi:10.1016/j.tet.2006.10.049 |
| 19. | Maury, J.; Feray, L.; Perfetti, P.; Bertrand, M. P. Org. Lett. 2010, 12, 3590–3593. doi:10.1021/ol101519w |
| 20. | Maury, J.; Mouysset, D.; Feray, L.; Marque, S. R. A.; Siri, D.; Bertrand, M. P. Chem.–Eur. J. 2012, 18, 3241–3247. doi:10.1002/chem.201102366 |
| 21. | Van der Deen, H.; Kellog, R. M.; Feringa, B. L. Org. Lett. 2000, 2, 1593–1595. doi:10.1021/ol005843+ |
| 22. | Miyabe, H.; Asada, R.; Yoshida, K.; Takemoto, Y. Synlett 2004, 540–542. doi:10.1055/s-2004-815407 |
| 23. | Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron 2005, 61, 385–393. doi:10.1016/j.tet.2004.10.104 |
| 24. | Yamada, K.-i.; Umeki, M.; Maekawa, M.; Yamamoto, Y.; Akindele, T.; Nakano, M.; Tomioka, K. Tetrahedron 2008, 64, 7258–7265. doi:10.1016/j.tet.2008.05.069 |
| 25. | Yamada, K.-i.; Konishi, T.; Nakano, M.; Fujii, S.; Cadou, R.; Yamamoto, Y.; Tomioka, K. J. Org. Chem. 2012, 77, 5775–5780. doi:10.1021/jo300944f |
| 26. | Cozzi, P. G. Angew. Chem., Int. Ed. 2006, 45, 2951–2954. doi:10.1002/anie.200504239 |
| 27. | Cozzi, P. G. Adv. Synth. Catal. 2006, 348, 2075–2079. doi:10.1002/adsc.200606178 |
| 28. | Cozzi, P. G.; Mignogna, A.; Vicennati, P. Adv. Synth. Catal. 2008, 350, 975–978. doi:10.1002/adsc.200700572 |
| 29. | Fernández-Ibáñez, M. A.; Maciá, B.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 1317–1319. doi:10.1002/anie.200704841 |
| 30. | Cozzi, P. G.; Benfatti, F.; Guiteras Capdevila, M.; Mignogna, A. Chem. Commun. 2008, 3317–3318. doi:10.1039/b805197f |
| 48. | Harada, T.; Katsuhira, T.; Oku, A. J. Org. Chem. 1992, 57, 5805–5807. doi:10.1021/jo00048a002 |
| 49. | Harada, T.; Katsuhira, T.; Hara, D.; Kotani, Y.; Maejima, K.; Kaji, R.; Oku, A. J. Org. Chem. 1993, 58, 4897–4907. doi:10.1021/jo00070a027 |
| 50. | Harada, T.; Katsuhira, T.; Hattori, K.; Oku, A. Tetrahedron 1994, 50, 7987–8002. doi:10.1016/S0040-4020(01)85284-4 |
| 5. | Lewiński, J.; Marciniac, W.; Lipkowski, J.; Justyniak, I. J. Am. Chem. Soc. 2003, 125, 12698–12699. doi:10.1021/ja036020t |
| 6. | Lewiński, J.; Śliwiński, W.; Dranka, M.; Justyniak, I.; Lipkowski, J. Angew. Chem., Int. Ed. 2006, 45, 4826–4829. doi:10.1002/anie.200601001 |
| 7. | Lewiński, J.; Suwała, K.; Kubisiak, M.; Ochal, Z.; Justyniak, I.; Lipkowski, J. Angew. Chem., Int. Ed. 2008, 47, 7888–7889. doi:10.1002/anie.200803254 |
| 8. | Lewiński, J.; Bury, W.; Dutkiewicz, M.; Maurin, M.; Justyniak, I.; Lipkowski, J. Angew. Chem., Int. Ed. 2008, 47, 573–576. doi:10.1002/anie.200703125 |
| 9. | Lewiński, J.; Kościelski, M.; Suwała, K.; Justyniak, I. Angew. Chem., Int. Ed. 2009, 48, 7017–7020. doi:10.1002/anie.200902716 |
| 10. | Lewiński, J.; Suwała, K.; Kaczorowski, T.; Gałęzowski, M.; Gryko, D. T.; Justyniak, I.; Lipkowski, J. Chem. Commun. 2009, 215–217. doi:10.1039/b813315h |
| 11. | Jana, S.; Berger, R. J. F.; Fröhlich, R.; Pape, T.; Mitzel, N. W. Inorg. Chem. 2007, 46, 4293–4297. doi:10.1021/ic062438r |
| 12. | Mileo, E.; Benfatti, F.; Cozzi, P. G.; Lucarini, M. Chem. Commun. 2009, 469–470. doi:10.1039/b818437b |
| 13. | Maury, J.; Feray, L.; Bazin, S.; Clément, J.-L.; Marque, S. R. A.; Siri, D.; Bertrand, M. P. Chem.–Eur. J. 2011, 17, 1586–1595. doi:10.1002/chem.201002616 |
| 14. | Mukherjee, D.; Ellern, A.; Sadow, A. D. J. Am. Chem. Soc. 2012, 134, 13018–13026. doi:10.1021/ja303440n |
| 38. | Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993 |
| 40. | Maury, J.; Feray, L.; Bertrand, M. P. Org. Lett. 2011, 13, 1884–1887. doi:10.1021/ol200419x |
| 38. | Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993 |
| 4. | Cohen, T.; Gibney, H.; Ivanov, R.; Yeh, E. A.-H.; Marek, I.; Curran, D. P. J. Am. Chem. Soc. 2007, 129, 15405–15409. doi:10.1021/ja076554k |
| 41. | Stüdemann, T.; Knochel, P. Angew. Chem., Int. Ed. Engl. 1997, 36, 93–95. doi:10.1002/anie.199700931 |
| 42. | Stüdemann, T.; Ibrahim-Ouali, M.; Knochel, P. Tetrahedron 1998, 54, 1299–1316. doi:10.1016/S0040-4020(97)10226-5 |
| 43. | Gourdet, B.; Lam, H. W. J. Am. Chem. Soc. 2009, 131, 3802–3803. doi:10.1021/ja900946h |
| 44. | Normant, J. F.; Alexakis, A. Synthesis 1981, 841–870. doi:10.1055/s-1981-29622 |
| 45. | Chinkov, N.; Tene, D.; Marek, I. In Metal-Catalyzed Cross Coupling Reactions, 2nd ed.; Diederich, D.; de Meijere, A., Eds.; Wiley-VCH: New York, 2004; p 395. |
| 46. | Denès, F.; Pérez-Luna, A.; Chemla, F. Chem. Rev. 2010, 110, 2366–2447. doi:10.1021/cr800420x |
| 4. | Cohen, T.; Gibney, H.; Ivanov, R.; Yeh, E. A.-H.; Marek, I.; Curran, D. P. J. Am. Chem. Soc. 2007, 129, 15405–15409. doi:10.1021/ja076554k |
| 39. | Chen, Z.; Zhang, Y.-X.; An, Y.; Song, X.-L.; Wang, Y.-H.; Zhu, L.-L.; Guo, L. Eur. J. Org. Chem. 2009, 5146–5152. doi:10.1002/ejoc.200900858 |
| 47. | Marek, I. Chem. Rev. 2000, 100, 2887–2900. doi:10.1021/cr990288e |
| 48. | Harada, T.; Katsuhira, T.; Oku, A. J. Org. Chem. 1992, 57, 5805–5807. doi:10.1021/jo00048a002 |
| 49. | Harada, T.; Katsuhira, T.; Hara, D.; Kotani, Y.; Maejima, K.; Kaji, R.; Oku, A. J. Org. Chem. 1993, 58, 4897–4907. doi:10.1021/jo00070a027 |
| 50. | Harada, T.; Katsuhira, T.; Hattori, K.; Oku, A. Tetrahedron 1994, 50, 7987–8002. doi:10.1016/S0040-4020(01)85284-4 |
| 51. | Creton, I.; Marek, I.; Normant, J.-F. Synthesis 1996, 1499–1508. doi:10.1055/s-1996-4422 |
| 36. | Feray, L.; Bertrand, M. P. Eur. J. Org. Chem. 2008, 3164–3170. doi:10.1002/ejoc.200800242 |
| 40. | Maury, J.; Feray, L.; Bertrand, M. P. Org. Lett. 2011, 13, 1884–1887. doi:10.1021/ol200419x |
| 37. | Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Chem.–Eur. J. 2008, 14, 8784–8788. doi:10.1002/chem.200801451 |
| 35. | Giboulot, S.; Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Tetrahedron Lett. 2008, 49, 3963–3966. doi:10.1016/j.tetlet.2008.04.108 |
| 61. | The stereochemical assignment was done on the basis of the comparison of the NMR data of 4ac and 4aa. Among others, the chemical shift (δ = 6.17 ppm) and coupling constant of the vinylic proton (J = 1.9 Hz) are indicative of the E-configuration. |
| 32. | Denes, F.; Chemla, F.; Normant, J. F. Angew. Chem., Int. Ed. 2003, 42, 4043–4046. doi:10.1002/anie.200250474 |
| 33. | Denes, F.; Cutri, S.; Pérez-Luna, A.; Chemla, F. Chem.–Eur. J. 2006, 12, 6506–6513. doi:10.1002/chem.200600334 |
| 34. | Denes, F.; Pérez-Luna, A.; Chemla, F. J. Org. Chem. 2007, 72, 398–406. doi:10.1021/jo061603h |
| 37. | Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Chem.–Eur. J. 2008, 14, 8784–8788. doi:10.1002/chem.200801451 |
| 38. | Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993 |
| 47. | Marek, I. Chem. Rev. 2000, 100, 2887–2900. doi:10.1021/cr990288e |
| 48. | Harada, T.; Katsuhira, T.; Oku, A. J. Org. Chem. 1992, 57, 5805–5807. doi:10.1021/jo00048a002 |
| 49. | Harada, T.; Katsuhira, T.; Hara, D.; Kotani, Y.; Maejima, K.; Kaji, R.; Oku, A. J. Org. Chem. 1993, 58, 4897–4907. doi:10.1021/jo00070a027 |
| 50. | Harada, T.; Katsuhira, T.; Hattori, K.; Oku, A. Tetrahedron 1994, 50, 7987–8002. doi:10.1016/S0040-4020(01)85284-4 |
| 51. | Creton, I.; Marek, I.; Normant, J.-F. Synthesis 1996, 1499–1508. doi:10.1055/s-1996-4422 |
| 37. | Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Chem.–Eur. J. 2008, 14, 8784–8788. doi:10.1002/chem.200801451 |
| 38. | Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993 |
| 51. | Creton, I.; Marek, I.; Normant, J.-F. Synthesis 1996, 1499–1508. doi:10.1055/s-1996-4422 |
| 52. |
Deng, C.-L.; Song, R.-J.; Liu, Y.-L.; Li, J.-H. Adv. Synth. Catal. 2009, 351, 3096–3100. doi:10.1002/adsc.200900588
The ZnCl2 catalyzed Conia–ene reaction of iodoalkynes that in principle involves a vinylidene zinc carbenoid intermediate has been reported here. |
| 55. | Creton, I.; Rezaei, H.; Marek, I.; Normant, J. F. Tetrahedron Lett. 1999, 40, 1899–1902. doi:10.1016/S0040-4039(99)00090-8 |
| 56. | Rezaei, H.; Marek, I.; Normant, J. F. Tetrahedron 2001, 57, 2477–2483. doi:10.1016/S0040-4020(01)00069-2 |
| 57. | Rezaei, H.; Yamanoi, S.; Chemla, F.; Normant, J. F. Org. Lett. 2000, 2, 419–421. doi:10.1021/ol991117z |
| 58. | Fallis, A. G.; Forgione, P. Tetrahedron 2001, 57, 5899–5913. doi:10.1016/S0040-4020(01)00422-7 |
| 37. | Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Chem.–Eur. J. 2008, 14, 8784–8788. doi:10.1002/chem.200801451 |
| 54. | Yeh, M. C. P.; Knochel, P. Tetrahedron Lett. 1989, 30, 4799–4802. doi:10.1016/S0040-4039(01)80511-6 |
| 48. | Harada, T.; Katsuhira, T.; Oku, A. J. Org. Chem. 1992, 57, 5805–5807. doi:10.1021/jo00048a002 |
| 49. | Harada, T.; Katsuhira, T.; Hara, D.; Kotani, Y.; Maejima, K.; Kaji, R.; Oku, A. J. Org. Chem. 1993, 58, 4897–4907. doi:10.1021/jo00070a027 |
| 50. | Harada, T.; Katsuhira, T.; Hattori, K.; Oku, A. Tetrahedron 1994, 50, 7987–8002. doi:10.1016/S0040-4020(01)85284-4 |
| 51. | Creton, I.; Marek, I.; Normant, J.-F. Synthesis 1996, 1499–1508. doi:10.1055/s-1996-4422 |
| 48. | Harada, T.; Katsuhira, T.; Oku, A. J. Org. Chem. 1992, 57, 5805–5807. doi:10.1021/jo00048a002 |
| 49. | Harada, T.; Katsuhira, T.; Hara, D.; Kotani, Y.; Maejima, K.; Kaji, R.; Oku, A. J. Org. Chem. 1993, 58, 4897–4907. doi:10.1021/jo00070a027 |
| 50. | Harada, T.; Katsuhira, T.; Hattori, K.; Oku, A. Tetrahedron 1994, 50, 7987–8002. doi:10.1016/S0040-4020(01)85284-4 |
| 51. | Creton, I.; Marek, I.; Normant, J.-F. Synthesis 1996, 1499–1508. doi:10.1055/s-1996-4422 |
| 53. | The Z configuration for the major isomer was determined by NOE experiments on isolated pure (Z)-4aa. An NOE signal (3%) was observed between the vinylic proton and the protons of the methylene group of the propyl chain α to the ester. |
| 37. | Pérez-Luna, A.; Botuha, C.; Ferreira, F.; Chemla, F. Chem.–Eur. J. 2008, 14, 8784–8788. doi:10.1002/chem.200801451 |
| 38. | Chemla, F.; Dulong, F.; Ferreira, F.; Nüllen, M.; Pérez-Luna, A. Synthesis 2011, 1347–1360. doi:10.1055/s-0030-1259993 |
© 2013 Chemla et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)