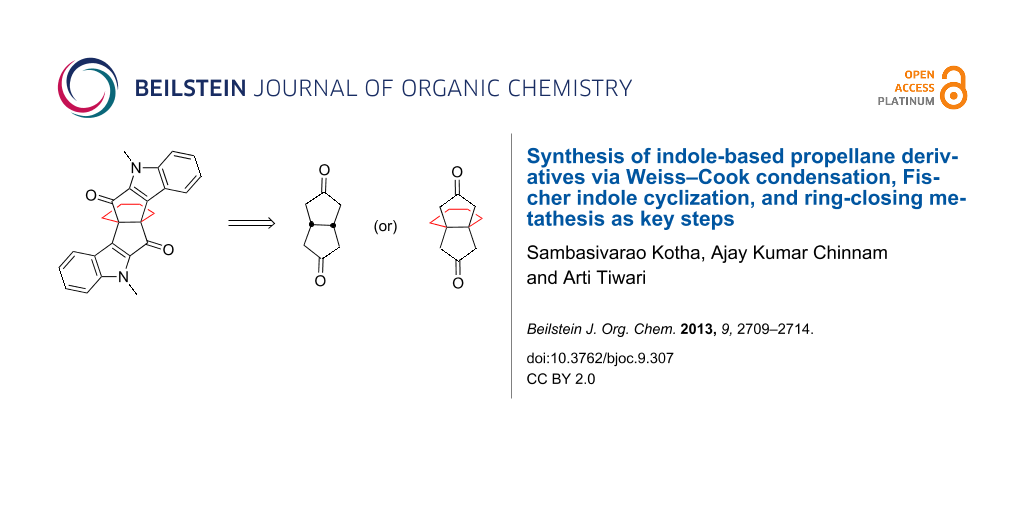
Abstract
A variety of highly functionalized indole-based [n.3.3]propellane derivatives is described. The synthesis of the propellane derivatives involves a Weiss–Cook condensation, a Fischer indole cyclization, and a ring-closing metathesis as key steps.

Graphical Abstract
Introduction
Propellanes are tricyclic systems conjoined with carbon–carbon single bonds (Figure 1) [1-3], and they are found to be highly congested. Some of these compounds are considered to be unstable entities and subjected to theoretical as well as synthetic studies [4]. Surprisingly, propellanes with larger rings, isolated from natural resources are found to be stable [5]. Among them, indole-based propellanes are useful in biology and medicine [6-12]. In 1963 Djerassi [13,14] isolated fendleridine and 1-acetylaspidoalbidine both of which belong to the aspidoalbine alkaloid family [15-20]. Kopsia alkaloids (Figure 2) show a wide range of structural diversity and exhibit interesting pharmacological activities [21]. For example, they are used for various ailments such as rheumatoid arthritis, edema, tonsillitis and hypertension. Lapidilectine B, grandilodine C [22], and lundurine B exhibit multidrug resistance (MDR) in vincristine-resistant KB cancer cells [23,24]. Minfiensine alkaloid [25-31] containing a novel 1,2,3,4-tetrahydrocarbazole ring skeleton shows anticancer activity [32]. König and co-workers identified some propellane derivatives as cannabinoid CB1 receptor antagonists [33], which are potential drugs for the treatment of schizophrenia and alcohol addiction [34]. However, the synthesis of indole alkaloid derivatives containing a propellane ring system is a challenging task due to the presence of quaternary centers involved with these systems [35].
![[1860-5397-9-307-1]](/bjoc/content/figures/1860-5397-9-307-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: [4.4.2] and [1.1.1]propellanes.
Figure 1: [4.4.2] and [1.1.1]propellanes.
![[1860-5397-9-307-2]](/bjoc/content/figures/1860-5397-9-307-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Alkaloids containing indole-based propellanes.
Figure 2: Alkaloids containing indole-based propellanes.
We envisioned a new synthetic route to diindole based propellane derivatives involving the Weiss–Cook condensation [36], Fischer indole cyclization [37,38], and ring-closing metathesis as key steps [39-42] (Figure 3).
![[1860-5397-9-307-3]](/bjoc/content/figures/1860-5397-9-307-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Retrosynthetic strategy to indole-based propellane 4.
Figure 3: Retrosynthetic strategy to indole-based propellane 4.
Here, we report a new synthetic strategy to indole-based propellane derivatives and our approach has several points of diversification: (i) various aryl and heteroaryl fused indole derivatives can be assembled by choosing an appropriate hydrazine derivative, (ii) during the alkylation of diketone 2 [43] various unsaturated alkenyl fragments may be incorporated either in a symmetrical or in an unsymmetrical manner, (iii) various functionalized cis-bicyclo[3.3.0]octane-3,7-dione derivatives are available by the Weiss–Cook reaction, (iv) the double bond generated at the end of the RCM sequence provides an additional handle for further synthetic manipulation.
Results and Discussion
To realize the strategy shown in Figure 3, cis-bicyclo[3.3.0]octane-3,7-dione (1) [44-50] was subjected to twofold Fischer indole cyclization to generate the diindole derivative 6 by using 1-methy-1-phenylhydrazine (5) under HCl/EtOH reflux conditions. Next, SeO2 oxidation of 6 in 1,4-dioxane under reflux gave the known diketone 2 (Scheme 1).
![[1860-5397-9-307-i1]](/bjoc/content/inline/1860-5397-9-307-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of diindole dione 2.
Scheme 1: Preparation of diindole dione 2.
Later, diketone 2 was treated with allyl bromide in the presence of NaH to afford the mono-allylated product 7 in 65% yield. The allyl group attacks the molecule from the sterically less hindered convex side. Since the alkylation step can be performed stepwise, symmetrical as well as unsymmetrical diindole derivatives can be generated. Therefore, mono-allyl derivative 7 was subjected to a second allylation with allyl bromide to generate the diallyl diketone 3 (86%) (Scheme 2). The second allyl group is also placed from the convex side of the molecule. Later, the mono-allyl diketone 7 was treated with 5-bromo-1-pentene in the presence of NaH to generate the unsymmetrical diketone 8.
![[1860-5397-9-307-i2]](/bjoc/content/inline/1860-5397-9-307-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of allylated indole derivatives 3, 7 and 8.
Scheme 2: Synthesis of allylated indole derivatives 3, 7 and 8.
Having compound 3 in hand, our next task was to construct the propellane derivatives via RCM by using Grubbs catalyst. In this regard, compound 3 was subjected to RCM under the influence of Grubbs 2nd generation catalyst in dry CH2Cl2 to furnish the desired RCM product 9 in 94% yield. The unsaturated propellane derivative 9 was subjected to hydrogenation in the presence of 10% Pd/C in dry EtOAc under H2 atmosphere to afford the saturated propellane derivative 4 in 95% yield (Scheme 3). Along similar lines, propellane derivative 10 was synthesized from unsymmetrical diketone 8 by using Grubbs 2nd generation catalyst and further subjected to hydrogenation to generate [6.3.3]propellane derivative 11. It is noteworthy that the formation of the eight membered ring is generally considered a difficult task due to unfavourable entropy factors [51-53], but in our strategy the eight membered ring is successfully assembled with the aid of Grubbs 2nd catalyst.
![[1860-5397-9-307-i3]](/bjoc/content/inline/1860-5397-9-307-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of indole-based propellane derivatives 4 and 11 by RCM route.
Scheme 3: Synthesis of indole-based propellane derivatives 4 and 11 by RCM route.
Diindole derivative 4 was also synthesized by an independent route starting with [4.3.3]propellane derivative 12 obtained by Weiss–Cook condensation with cyclohexane-1,2-dione. Later, the propellane 12 was subjected to a twofold Fischer indole cyclization with 1-methyl-1-phenylhydrazine (5) with SOCl2/EtOH under reflux to deliver the diindole derivative 13 in 34% yield [54]. The diindole derivative 13 was subjected to SeO2 oxidation to generate the diketone 4 in 76% yield. The spectral data of the compound 4 obtained by this route (Scheme 4) is found to be identical with that of the compound obtained by the earlier route.
![[1860-5397-9-307-i4]](/bjoc/content/inline/1860-5397-9-307-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 4 by Weiss–Cook condensation and two fold Fischer indole cyclization.
Scheme 4: Synthesis of 4 by Weiss–Cook condensation and two fold Fischer indole cyclization.
Conclusion
We have developed a new and useful synthetic strategy to indole-based propellane derivatives with simple starting materials involving RCM as a key step. The structure of compound 4 has been elucidated based on spectral data and additionally by an independent synthetic sequence.
Experimental
NMR spectra were recorded at rt on a 400 MHz Bruker NMR spectrometer in CDCl3 solution. Coupling constants (J values) are given in Hertz (Hz). Melting points were recorded with a Büchi melting point apparatus. Infrared (IR) spectra were recorded by using a Nicolet Impact-400 FTIR spectrometer in KBr. The high-resolution mass spectrometric measurements were carried out with a Micromass Q-ToF spectrometer. Analytical thin-layer chromatography (TLC) was performed on (10 × 5 cm) glass plates coated with Acme’s silica gel GF254 (containing 13% calcium sulfate as a binder). Chromatography was performed with Acme’s silica gel (100–200 mesh) by using double spray bellows for the application of pressure, and the column was eluted with an ethyl acetate/petroleum ether mixture. The organic solvents used in this study were dried over appropriate drying agents and distilled prior to use.
6a-Allyl-cis-5,6a,11,12a-tetrahydro-5,11-dimethylpentaleno[2,1-b:5,4-b’]diindole-6,12-dione (7): To a suspension of NaH (1.25 mmol) in THF (10 mL), diketone 2 (100 mg, 0.3 mmol) was added at room temperature under a nitrogen atmosphere. Then, the resulting reaction mixture was heated up to 65 °C for 15 min. After cooling to room temperature, allyl bromide (0.02 mL, 0.3 mmol) was added to the reaction mixture dropwise, and stirring was continued at room temperature for 2 h. At the end of the reaction (TLC monitoring), the reaction mixture was diluted with ethyl acetate (10 mL), washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product obtained was purified by silica gel column chromatography (5% EtOAc/petroleum ether) to give compound 7 (73 mg). Yellow colored solid; 65% yield. Rf = 0.42 (silica gel, 5% EtOAc/petroleum ether); mp: 176–178 °C; IR (KBr) νmax: 3023, 2986, 2934, 1735, 1447, 1248, 1048 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.04–3.18 (m, 2H), 3.82 (s, 3H), 3.85 (s, 3H), 4.53 (s, 1H), 5.02 (dd, J = 10.0, 1.5 Hz, 1H), 5.19 (dd, J = 16.9, 1.4 Hz, 1H), 5.66–5.73 (m, 1H), 7.22–7.29 (m, 2H), 7.31–7.39 (m, 2H), 7.40–7.44 (m, 2H), 8.06–8.10 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 30.39, 38.90, 58.73, 63.55, 111.18, 111.29, 119.24, 121.38, 121.40, 122.58, 123.09, 123.36, 123.51, 127.53, 127.64, 133.63, 134.92, 135.04, 138.90, 142.41, 144.94, 145.01, 189.86, 192.03; HRMS (ESI, Q-ToF) m/z: [M + H]+ calcd for C25H21N2O2, 381.1603; found, 381.1599.
6a,12a-Diallyl-cis-5,6a,11,12a-tetrahydro-5,11-dimethylpentaleno[2,1-b:5,4-b’]diindole-6,12-dione (3): To a suspension of NaH (1.25 mmol) in THF (10 mL), mono-allyl diketone 7 (70 mg, 0.18 mmol) was added at room temperature under a nitrogen atmosphere. Then, the resulting reaction mixture was heated up to 65 °C for 15 min. After cooling to room temperature, allyl bromide (0.02 mL, 0.27 mmol) was added to the reaction mixture in a dropwise manner, and stirring continued at room temperature for 6 h. At the end of the reaction (TLC monitoring), the reaction mixture was diluted with ethyl acetate (10 mL), washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product obtained was purified by silica gel column chromatography (5% EtOAc/petroleum ether) to deliver the compound 3 (67 mg). Yellow colored solid; 86% yield. Rf = 0.45 (silica gel, 5% EtOAc/petroleum ether); mp: 225–227 °C; IR (KBr) νmax: 2978, 2961, 2928, 1740, 1463, 1242, 1047 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.17–3.23 (m, 2H), 3.44–3.49 (m, 2H), 3.82 (s, 6H), 4.98 (dd, J = 10.2, 1.2 Hz, 2H), 5.19 (dd, J = 17.1, 1.2 Hz, 2H), 5.65–5.72 (m, 2H), 7.20–7.29 (m, 2H), 7.32–7.37 (m, 2H), 7.39–7.41 (m, 2H), 8.04 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 30.28, 35.45, 67.02, 111.05, 117.76, 121.14, 122.51, 123.50, 127.26, 133.49, 134.54, 142.22, 144.61, 192.08.; HRMS (ESI, Q-ToF) m/z: [M + H]+calcd for C28H25N2O2, 421.1916; found, 421.1917.
5,11-Dimethyl-6a,12a-but[2]enopentaleno[2,1-b:5,4-b']diindole-6,12(5H,11H)-dione (9): A solution of diallyl dione 3 (65 mg, 0.15 mmol) in dry CH2Cl2 (15 mL) was degassed with N2 for 10 min, then, Grubbs 2nd generation catalyst (12 mg, 0.014 mmol) was added at room temperature and stirred for 24 h. At the end of the reaction (TLC monitoring), the solvent was removed in vacuo and the crude product was purified by silica gel column chromatography (5% EtOAc/petroleum ether) to give compound 9 (57 mg). Colourless solid; 94% yield. Rf = 0.40 (silica gel, 5% EtOAc/petroleum ether); mp: 295–297 °C; IR (KBr) νmax: 3049, 2930, 1686, 1266, 1031 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.86 (dd, J = 14.8, 2.6 Hz, 2H), 3.10 (dd, J = 14.2, 1.9 Hz, 2H), 3.83 (s, 6H), 5.95 (dd, J = 12.0, 8.8 Hz, 2H), 7.22–7.24 (m, 2H), 7.30–7.32 (m, 2H), 7.38–7.42 (m, 2H), 8.04 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 30.41, 31.32, 64.23, 111.25, 121.26, 122.37, 123.37, 127.39, 128.25, 135.16, 142.35, 145.01, 192.57; HRMS (ESI, Q-ToF) m/z: [M + H]+ calcd for C26H21N2O2, 393.1603; found, 393.1609.
5,11-Dimethyl-6a,12a-butanopentaleno[2,1-b:5,4-b']diindole-6,12(5H,11H)-dione (4): To a solution of propellane 9 (50 mg, 0.12 mmol) in dry EtOAc (10 mL), 10% Pd/C (10 mg, 0.09 mmol) was added and the reaction mixture was stirred at room temperature under H2 atmosphere (1 atm) for 28 h. At the end of the reaction (TLC monitoring), the reaction mixture was filtered through a pad of celite and washed with ethyl acetate (20 mL). Evaporation of the solvent in vacuo gave the crude product. Further purification by silica-gel column chromatography (5% EtOAc/petroleum ether) gave the hydrogenated product 4 (48 mg). Colorless solid; 95% yield. Rf = 0.42 (silica gel, 5% EtOAc/petroleum ether); mp: 305–307 °C; IR (KBr) νmax: 2929, 2851, 1686, 1267, 1047 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.49–1.54 (m, 2H), 1.58–1.63 (m, 2H), 2.26–2.34 (m, 2H), 2.50–2.57 (m, 2H), 3.84 (s, 6H), 7.21–7.27 (m, 2H), 7.31–7.34 (m, 2H), 7.38–7.42 (m, 2H), 8.06 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 17.16, 28.07, 30.25, 63.11, 111.04, 121.00, 122.56, 123.30, 127.19, 134.76, 142.73, 144.80, 193.29; HRMS (ESI, Q-ToF) m/z: [M + H]+ calcd for C26H23N2O2, 395.1760; found, 395.1750.
To a solution of propellane 13 (33 mg, 0.09 mmol) in dioxane (10 mL), SeO2 (22 mg, 0.20 mmol) was added and the reaction mixture was refluxed for 3 h. At the end of the reaction (TLC monitoring), the reaction mixture was filtered through a pad of celite and washed with a 1:1 mixture of CCl4 and CHCl3 (20 mL). Evaporation of the solvent in vacuo gave the crude product, which was further purified by silica-gel column chromatography. Elution of the column with 5% EtOAc/petroleum ether gave the diketone 4 (27 mg, 76%) as a white solid. The spectral data of this compound is identical with that of compound 4 obtained by the other route.
Supporting Information
| Supporting Information File 1: Copies of 1H, 13C NMR and HRMS spectra for all new compounds. | ||
| Format: PDF | Size: 3.7 MB | Download |
Acknowledgements
We thank the Department of Science and Technology (DST), New Delhi for the financial support and the Sophisticated Analytical Instrument Facility (SAIF), IIT-Bombay for recording spectral data. S.K. thanks the Department of Science and Technology for the award of a J. C. Bose fellowship. A.K.C. thanks the University Grants Commission, New Delhi for the award of a research fellowship. A.T. thanks the CSIR for the award of a research fellowship.
References
-
Ginsburg, D. Propellanes Structure and Reactions; Verlag Chemie: Weinheim, 1975.
Return to citation in text: [1] -
Ginsburg, D. Propellanes Structure and Reactions (Sequel I); Department of Chemistry, Technion: Haifa, 1981.
Return to citation in text: [1] -
Ginsburg, D. Propellanes Structure and Reactions (Sequel II); Department of Chemistry, Technion: Haifa, 1985.
Return to citation in text: [1] -
Ginsburg, D. Top. Curr. Chem. 1987, 137, 1. doi:10.1007/3-540-16904-0_13
Return to citation in text: [1] -
Pihko, A. J.; Koskinen, A. M. P. Tetrahedron 2005, 61, 8769. doi:10.1016/j.tet.2005.06.013
Return to citation in text: [1] -
Sundberg, R. J. Indoles; Academic Press: San Diego, CA, 1996.
Return to citation in text: [1] -
Gilchrist, T. L. J. Chem. Soc., Perkin Trans. 1 1999, 2849. doi:10.1039/A808162J
Return to citation in text: [1] -
Gribble, G. W. J. Chem. Soc., Perkin Trans. 1 2000, 1045. doi:10.1039/A909834H
Return to citation in text: [1] -
Makosza, M.; Wojciechowski, K. Heterocycles 2001, 54, 445. doi:10.3987/REV-00-SR(I)2
Return to citation in text: [1] -
Battistuzzi, G.; Cacchi, S.; Fabrizi, G. Eur. J. Org. Chem. 2002, 2671. doi:10.1002/1099-0690(200208)2002:16<2671::AID-EJOC2671>3.0.CO;2-X
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105, 2873. doi:10.1021/cr040639b
Return to citation in text: [1] -
Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875. doi:10.1021/cr0505270
Return to citation in text: [1] -
Brown, K. S., Jr.; Budzikiewicz, H.; Djerassi, C. Tetrahedron Lett. 1963, 4, 1731. doi:10.1016/S0040-4039(01)90904-9
Return to citation in text: [1] -
Walser, A.; Djerassi, C. Helv. Chim. Acta 1965, 48, 391. doi:10.1002/hlca.19650480220
Return to citation in text: [1] -
Hesse, M. Indolalkaloide in Tabellen-Ergänzungswerk; Springer-Verlag: Berlin, 1968; p 77.
Return to citation in text: [1] -
Honma, Y.; Ohnuma, T.; Ban, Y. Heterocycles 1976, 5, 47. doi:10.3987/S-1976-01-0047
Return to citation in text: [1] -
Ban, Y.; Ohnuma, T.; Seki, K.; Oishi, T. Tetrahedron Lett. 1975, 16, 727. doi:10.1016/S0040-4039(00)71968-X
Return to citation in text: [1] -
Yoshida, K.; Sakuma, Y.; Ban, Y. Heterocycles 1987, 25, 47. doi:10.3987/S-1987-01-0047
Return to citation in text: [1] -
Campbell, E. L.; Zuhl, A. M.; Lui, C. M.; Boger, D. L. J. Am. Chem. Soc. 2010, 132, 3009. doi:10.1021/ja908819q
Return to citation in text: [1] -
Mitaine, A.-C.; Mesbah, K.; Richard, B.; Petermann, C.; Arrazola, S.; Moretti, C.; Zèches-Hanrot, M.; Le Men-Olivier, L. Planta Med. 1996, 62, 458. doi:10.1055/s-2006-957939
Return to citation in text: [1] -
Awang, K.; Sévenet, T.; Païs, M.; Hadi, H. A. J. Nat. Prod. 1993, 56, 1134. doi:10.1021/np50097a018
Return to citation in text: [1] -
Yap, W.-S.; Gan, C.-Y.; Low, Y.-Y.; Choo, Y.-M.; Etoh, T.; Hayashi, M.; Komiyama, K.; Kam, T.-S. J. Nat. Prod. 2011, 74, 1309. doi:10.1021/np200008g
Return to citation in text: [1] -
Kam, T.-S.; Lim, K.-H.; Yoganathan, K.; Hayashi, M.; Komiyama, K. Tetrahedron 2004, 60, 10739. doi:10.1016/j.tet.2004.08.091
Return to citation in text: [1] -
Schultz, E. E.; Pujanauski, B. G.; Sarpong, R. Org. Lett. 2012, 14, 648. doi:10.1021/ol203302f
Return to citation in text: [1] -
Massiot, G.; Thépenier, P.; Jacquier, M.-J.; Le Men-Olivier, L.; Delaude, C. Heterocycles 1989, 29, 1435. doi:10.3987/COM-89-4987
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E.; Wrobleski, A. D. J. Am. Chem. Soc. 2005, 127, 10186. doi:10.1021/ja0533895
Return to citation in text: [1] -
Dounay, A. B.; Humphreys, P. G.; Overman, L. E.; Wrobleski, A. D. J. Am. Chem. Soc. 2008, 130, 5368. doi:10.1021/ja800163v
Return to citation in text: [1] -
Shen, L.; Zhang, M.; Wu, Y.; Qin, Y. Angew. Chem., Int. Ed. 2008, 47, 3618. doi:10.1002/anie.200800566
Return to citation in text: [1] -
Jones, S. B.; Simmons, B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 13606. doi:10.1021/ja906472m
Return to citation in text: [1] -
Li, G.; Padwa, A. Org. Lett. 2011, 13, 3767. doi:10.1021/ol201320v
Return to citation in text: [1] -
Liu, P.; Wang, J.; Zhang, J.; Qiu, F. G. Org. Lett. 2011, 13, 6426. doi:10.1021/ol2027224
Return to citation in text: [1] -
Ramírez, A.; García-Rubio, S. Curr. Med. Chem. 2003, 10, 1891. doi:10.2174/0929867033457016
Return to citation in text: [1] -
Elsebai, M. F.; Rempel, V.; Schnakenburg, G.; Kehraus, S.; Müller, C. E.; König, G. M. ACS Med. Chem. Lett. 2011, 2, 866. doi:10.1021/ml200183z
Return to citation in text: [1] -
Pacher, P.; Bátkai, S.; Kunos, G. Pharmacol. Rev. 2006, 58, 389. doi:10.1124/pr.58.3.2
Return to citation in text: [1] -
Cox, E. D.; Cook, J. M. Chem. Rev. 1995, 95, 1797. doi:10.1021/cr00038a004
Return to citation in text: [1] -
Bertz, S. H.; Cook, J. M.; Gawish, A.; Weiss, U. Org. Synth. 1986, 64, 27.
Return to citation in text: [1] -
Gribble, G. W. Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Topics in Heterocyclic Chemistry; Springer: Berlin, Heidelberg, 2010. doi:10.1007/978-3-642-15733-2
Return to citation in text: [1] -
Inman, M.; Moody, C. J. Chem. Sci. 2013, 4, 29. doi:10.1039/c2sc21185h
Return to citation in text: [1] -
Kotha, S.; Dipak, M. K. Chem.–Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366
Return to citation in text: [1] -
Kotha, S.; Lahari, K. Synlett 2007, 2767. doi:10.1055/s-2007-990954
Return to citation in text: [1] -
Kotha, S.; Chavan, A. S.; Dipak, M. K. Tetrahedron 2011, 67, 501. doi:10.1016/j.tet.2010.10.080
Return to citation in text: [1] -
Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397. doi:10.1016/j.tet.2011.10.018
Return to citation in text: [1] -
Kotha, S.; Hollinshead, S.; Grubisha, D.; Laib, F.; Bennett, D.; Cook, J. M. J. Org. Chem. 1990, 55, 3858. doi:10.1021/jo00299a031
Return to citation in text: [1] -
Paquette, L. A.; Crouse, G. D. J. Org. Chem. 1983, 48, 141. doi:10.1021/jo00149a035
Return to citation in text: [1] -
Deshpande, M. N.; Jawdosiuk, M.; Kubiak, G.; Venkatachalam, M.; Weiss, U.; Cook, J. M. J. Am. Chem. Soc. 1985, 107, 4786. doi:10.1021/ja00302a035
Return to citation in text: [1] -
Fu, X.; Cook, J. M. J. Org. Chem. 1992, 57, 5121. doi:10.1021/jo00045a023
Return to citation in text: [1] -
Fu, X.; Cook, J. M. Aldrichimica Acta 1992, 25, 43.
Return to citation in text: [1] -
Gupta, A. K.; Fu, X.; Snyder, J. P.; Cook, J. M. Tetrahedron 1991, 47, 3665. doi:10.1016/S0040-4020(01)80896-6
Return to citation in text: [1] -
Weiss, U.; Edwards, J. M. Tetrahedron Lett. 1968, 9, 4885. doi:10.1016/S0040-4039(00)72784-5
Return to citation in text: [1] -
Yang, S.; Cook, J. M. J. Org. Chem. 1976, 41, 1903. doi:10.1021/jo00873a004
Return to citation in text: [1] -
Miller, S. J.; Kim, S.-H.; Chen, Z.-R.; Grubbs, R. H. J. Am. Chem. Soc. 1995, 117, 2108. doi:10.1021/ja00112a031
Return to citation in text: [1] -
Maier, M. E. Angew. Chem., Int. Ed. 2000, 39, 2073. doi:10.1002/1521-3773(20000616)39:12<2073::AID-ANIE2073>3.0.CO;2-0
Return to citation in text: [1] -
Tori, M.; Mizutani, R. Molecules 2010, 15, 4242. doi:10.3390/molecules15064242
Return to citation in text: [1] -
Kotha, S.; Chinnam, A. K. unpublished results.
Return to citation in text: [1]
| 44. | Paquette, L. A.; Crouse, G. D. J. Org. Chem. 1983, 48, 141. doi:10.1021/jo00149a035 |
| 45. | Deshpande, M. N.; Jawdosiuk, M.; Kubiak, G.; Venkatachalam, M.; Weiss, U.; Cook, J. M. J. Am. Chem. Soc. 1985, 107, 4786. doi:10.1021/ja00302a035 |
| 46. | Fu, X.; Cook, J. M. J. Org. Chem. 1992, 57, 5121. doi:10.1021/jo00045a023 |
| 47. | Fu, X.; Cook, J. M. Aldrichimica Acta 1992, 25, 43. |
| 48. | Gupta, A. K.; Fu, X.; Snyder, J. P.; Cook, J. M. Tetrahedron 1991, 47, 3665. doi:10.1016/S0040-4020(01)80896-6 |
| 49. | Weiss, U.; Edwards, J. M. Tetrahedron Lett. 1968, 9, 4885. doi:10.1016/S0040-4039(00)72784-5 |
| 50. | Yang, S.; Cook, J. M. J. Org. Chem. 1976, 41, 1903. doi:10.1021/jo00873a004 |
| 39. | Kotha, S.; Dipak, M. K. Chem.–Eur. J. 2006, 12, 4446. doi:10.1002/chem.200501366 |
| 40. | Kotha, S.; Lahari, K. Synlett 2007, 2767. doi:10.1055/s-2007-990954 |
| 41. | Kotha, S.; Chavan, A. S.; Dipak, M. K. Tetrahedron 2011, 67, 501. doi:10.1016/j.tet.2010.10.080 |
| 42. | Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397. doi:10.1016/j.tet.2011.10.018 |
| 43. | Kotha, S.; Hollinshead, S.; Grubisha, D.; Laib, F.; Bennett, D.; Cook, J. M. J. Org. Chem. 1990, 55, 3858. doi:10.1021/jo00299a031 |
| 1. | Ginsburg, D. Propellanes Structure and Reactions; Verlag Chemie: Weinheim, 1975. |
| 2. | Ginsburg, D. Propellanes Structure and Reactions (Sequel I); Department of Chemistry, Technion: Haifa, 1981. |
| 3. | Ginsburg, D. Propellanes Structure and Reactions (Sequel II); Department of Chemistry, Technion: Haifa, 1985. |
| 13. | Brown, K. S., Jr.; Budzikiewicz, H.; Djerassi, C. Tetrahedron Lett. 1963, 4, 1731. doi:10.1016/S0040-4039(01)90904-9 |
| 14. | Walser, A.; Djerassi, C. Helv. Chim. Acta 1965, 48, 391. doi:10.1002/hlca.19650480220 |
| 6. | Sundberg, R. J. Indoles; Academic Press: San Diego, CA, 1996. |
| 7. | Gilchrist, T. L. J. Chem. Soc., Perkin Trans. 1 1999, 2849. doi:10.1039/A808162J |
| 8. | Gribble, G. W. J. Chem. Soc., Perkin Trans. 1 2000, 1045. doi:10.1039/A909834H |
| 9. | Makosza, M.; Wojciechowski, K. Heterocycles 2001, 54, 445. doi:10.3987/REV-00-SR(I)2 |
| 10. | Battistuzzi, G.; Cacchi, S.; Fabrizi, G. Eur. J. Org. Chem. 2002, 2671. doi:10.1002/1099-0690(200208)2002:16<2671::AID-EJOC2671>3.0.CO;2-X |
| 11. | Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105, 2873. doi:10.1021/cr040639b |
| 12. | Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875. doi:10.1021/cr0505270 |
| 37. | Gribble, G. W. Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Topics in Heterocyclic Chemistry; Springer: Berlin, Heidelberg, 2010. doi:10.1007/978-3-642-15733-2 |
| 38. | Inman, M.; Moody, C. J. Chem. Sci. 2013, 4, 29. doi:10.1039/c2sc21185h |
| 5. | Pihko, A. J.; Koskinen, A. M. P. Tetrahedron 2005, 61, 8769. doi:10.1016/j.tet.2005.06.013 |
| 34. | Pacher, P.; Bátkai, S.; Kunos, G. Pharmacol. Rev. 2006, 58, 389. doi:10.1124/pr.58.3.2 |
| 23. | Kam, T.-S.; Lim, K.-H.; Yoganathan, K.; Hayashi, M.; Komiyama, K. Tetrahedron 2004, 60, 10739. doi:10.1016/j.tet.2004.08.091 |
| 24. | Schultz, E. E.; Pujanauski, B. G.; Sarpong, R. Org. Lett. 2012, 14, 648. doi:10.1021/ol203302f |
| 32. | Ramírez, A.; García-Rubio, S. Curr. Med. Chem. 2003, 10, 1891. doi:10.2174/0929867033457016 |
| 22. | Yap, W.-S.; Gan, C.-Y.; Low, Y.-Y.; Choo, Y.-M.; Etoh, T.; Hayashi, M.; Komiyama, K.; Kam, T.-S. J. Nat. Prod. 2011, 74, 1309. doi:10.1021/np200008g |
| 33. | Elsebai, M. F.; Rempel, V.; Schnakenburg, G.; Kehraus, S.; Müller, C. E.; König, G. M. ACS Med. Chem. Lett. 2011, 2, 866. doi:10.1021/ml200183z |
| 21. | Awang, K.; Sévenet, T.; Païs, M.; Hadi, H. A. J. Nat. Prod. 1993, 56, 1134. doi:10.1021/np50097a018 |
| 51. | Miller, S. J.; Kim, S.-H.; Chen, Z.-R.; Grubbs, R. H. J. Am. Chem. Soc. 1995, 117, 2108. doi:10.1021/ja00112a031 |
| 52. | Maier, M. E. Angew. Chem., Int. Ed. 2000, 39, 2073. doi:10.1002/1521-3773(20000616)39:12<2073::AID-ANIE2073>3.0.CO;2-0 |
| 53. | Tori, M.; Mizutani, R. Molecules 2010, 15, 4242. doi:10.3390/molecules15064242 |
| 15. | Hesse, M. Indolalkaloide in Tabellen-Ergänzungswerk; Springer-Verlag: Berlin, 1968; p 77. |
| 16. | Honma, Y.; Ohnuma, T.; Ban, Y. Heterocycles 1976, 5, 47. doi:10.3987/S-1976-01-0047 |
| 17. | Ban, Y.; Ohnuma, T.; Seki, K.; Oishi, T. Tetrahedron Lett. 1975, 16, 727. doi:10.1016/S0040-4039(00)71968-X |
| 18. | Yoshida, K.; Sakuma, Y.; Ban, Y. Heterocycles 1987, 25, 47. doi:10.3987/S-1987-01-0047 |
| 19. | Campbell, E. L.; Zuhl, A. M.; Lui, C. M.; Boger, D. L. J. Am. Chem. Soc. 2010, 132, 3009. doi:10.1021/ja908819q |
| 20. | Mitaine, A.-C.; Mesbah, K.; Richard, B.; Petermann, C.; Arrazola, S.; Moretti, C.; Zèches-Hanrot, M.; Le Men-Olivier, L. Planta Med. 1996, 62, 458. doi:10.1055/s-2006-957939 |
| 25. | Massiot, G.; Thépenier, P.; Jacquier, M.-J.; Le Men-Olivier, L.; Delaude, C. Heterocycles 1989, 29, 1435. doi:10.3987/COM-89-4987 |
| 26. | Dounay, A. B.; Overman, L. E.; Wrobleski, A. D. J. Am. Chem. Soc. 2005, 127, 10186. doi:10.1021/ja0533895 |
| 27. | Dounay, A. B.; Humphreys, P. G.; Overman, L. E.; Wrobleski, A. D. J. Am. Chem. Soc. 2008, 130, 5368. doi:10.1021/ja800163v |
| 28. | Shen, L.; Zhang, M.; Wu, Y.; Qin, Y. Angew. Chem., Int. Ed. 2008, 47, 3618. doi:10.1002/anie.200800566 |
| 29. | Jones, S. B.; Simmons, B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 13606. doi:10.1021/ja906472m |
| 30. | Li, G.; Padwa, A. Org. Lett. 2011, 13, 3767. doi:10.1021/ol201320v |
| 31. | Liu, P.; Wang, J.; Zhang, J.; Qiu, F. G. Org. Lett. 2011, 13, 6426. doi:10.1021/ol2027224 |
© 2013 Kotha et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)