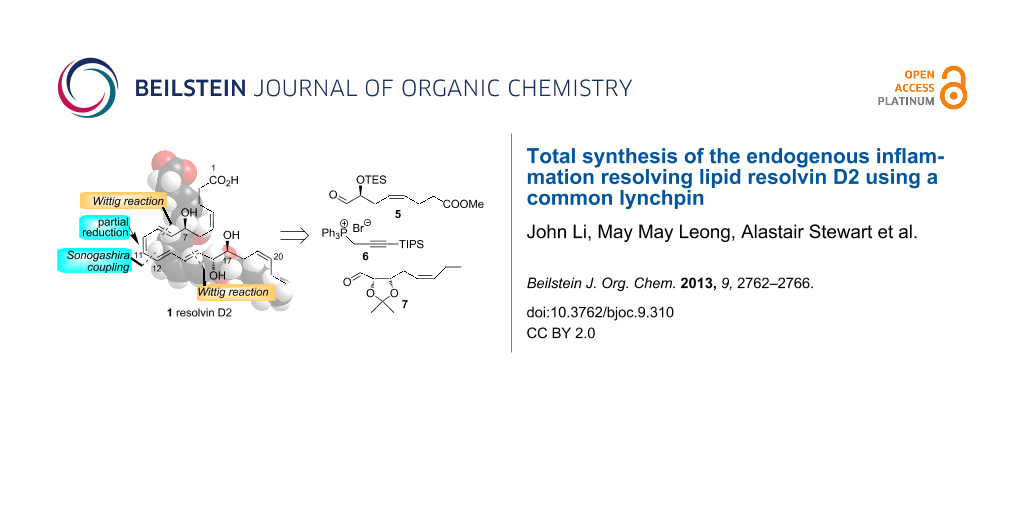
Abstract
The total synthesis of the endogenous inflammation resolving eicosanoid resolvin D2 (1) is described. The key steps involved a Wittig reaction between aldehyde 5 and the ylide derived from phosphonium salt 6 to give enyne 17 and condensation of the same ylide with aldehyde 7 to afford enyne 11. Desilylation of 11 followed by hydrozirconation and iodination gave the vinyl iodide 4 and Sonogashira coupling between this compound and enyne 3 provided alkyne 18. Acetonide deprotection, partial reduction and ester hydrolysis then gave resolvin D2 (1).

Graphical Abstract
Introduction
The resolution of inflammation is a tightly governed active process effectively mediated by a range of bioactive polyunsaturated fatty acids, peptides and proteins. In 2002, a new family of endogenously generated lipid mediators involved in the resolution of inflammation named the resolvins (resolution phase interaction products) were identified by Serhan and co-workers in the inflammatory exudates of aspirin treated mice [1-3]. The resolvins are divided into 2 groups, the D-series resolvins D2 (1) and D1 (2) [3], which are derivatives of docosahexaenoic acid (DHA) (Figure 1) and the E-series [4] derived from eicosapentaenoic acid. Structural analysis by mass spectrometry (MS) showed that resolvin D2 (RvD2, 1) was a 17-hydroxy derivative of DHA (17HDHA) [1]. However, no NMR experiments were performed due to nanogram quantities isolated and the stereochemistry was tentatively assigned based on the proposed biosynthesis via lipoxygenase modification of DHA.
![[1860-5397-9-310-1]](/bjoc/content/figures/1860-5397-9-310-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of resolvins D1 (1) and D2 (2).
Figure 1: Structures of resolvins D1 (1) and D2 (2).
RvD2 (1) prevents the adherence of polymorphonuclear leukocytes (PMN) to the blood vessel wall by promoting the shedding of L-selectin from PMNs thus preventing binding to E selectin on the endothelial cell lining of the blood vessel [5]. Furthermore, RvD2 (1) promotes the influx and phagocytic activity of macrophages, facilitating clearance of dead cells and microbial pathogens, allowing resolution of inflammation and infection [5]. This successful evaluation of the resolvin series in preclinical models of bacterial sepsis has stimulated strong interest in their therapeutic potential, as RvD2 appears to express the unusual combination of anti-inflammatory and antimicrobial activity. Further interest in inflammation-resolving lipids is stimulated by their inhibitory effects on inflammatory pain, which are mediated via inhibition of the activity of TRPV1 and TRPA1 calcium channels on sensory nerves [6]. Resolvin D1 (2) [7] has been shown to act directly on human PMNs and also regulates actin polymerization [8]. Whilst RvD1 has been shown to act on the FPR2 and GPR32 types of G-Protein-coupled receptors, the receptor(s) for RvD2 remain to be identified. Identification of the receptors mediating the combined anti-inflammatory and antimicrobial actions would facilitate efforts to identify ligands that have better drug-like properties than RvD2 or its analogues. However, such efforts have been limited by the lack of availability of suitable amounts of RvD2.
The first total synthesis of (7S,16R,17S)-RvD2 (1) was communicated by Spur in 2004 [9] but this report did not include an experimental section although physical data for some compounds was provided. A similar synthesis of RvD2 was utilized by others for the production of 1 for a biological study [5] but again, there was no experimental provided. The total synthesis of resolvin D1 has also been reported [10] along with resolvins D3 [11], D5 [12], D6 [13] and resolvins E1 [4,14,15], E2 [16,17] and E3 [18] with full experimental details included for resolvins D3 [11], E2 [16] and E3 [18]. An improved synthesis of the C16–C20 fragment of resolvin E1 has also been reported [19]. We were interested in accessing amounts of RvD2 (1) for biological evaluation but without detailed synthetic sequence to follow and given the very high cost [20] of commercial 1 we elected to develop an alternative route to provide this important compound and analogues for further biological evaluation. Herein we describe a synthesis of RvD2 (1) which includes full experimental details so that other researchers can produce useful amounts of this important compound as well as novel isomers.
Results and Discussion
Retrosynthetic analysis
A retrosynthetic analysis of RvD2 (1) is shown in Scheme 1. It was envisaged that the target compound 1 could be secured via a Sonogashira coupling to form the C11–C12 bond followed by partial reduction. This is similar to the endgame of the reported syntheses of 1 [5,9] but both of these approaches involved formation of the C9–C10 bond as the convergent step. In our approach, 1 could arise from enyne 3 and vinyl iodide 4 which could both be obtained by Wittig extension using the common linchpin phosphorus ylide derived from phosphonium salt 6 [21,22] and each of the homochiral aldehydes 5 and 7 [9].
![[1860-5397-9-310-i1]](/bjoc/content/inline/1860-5397-9-310-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of RvD2 (1).
Scheme 1: Retrosynthetic analysis of RvD2 (1).
Synthesis of vinyl iodide 4
The synthesis of fragment 4 began with the production of the aldehyde 7 as shown in Scheme 2. A Wittig reaction between hemiacetal 8 [23] and the ylide derived from 9 provided the alkene 10 [9] with excellent stereoselectivity. Oxidation of 10 with Dess–Martin periodinane then afforded aldehyde 7. The phosphonium salt 6 [21,22] was produced from propargyl bromide via silylation of the derived sodium salt with TIPSCl followed by reaction with triphenylphosphine.
![[1860-5397-9-310-i2]](/bjoc/content/inline/1860-5397-9-310-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of aldehyde 7 and phosphonium salt 6.
Scheme 2: Synthesis of aldehyde 7 and phosphonium salt 6.
Treatment of the salt 6 with n-BuLi gave the ylide and condensation with the aldehyde 7 afforded the desired E-enyne 11 along with the Z-isomer in a ratio of 2.2:1 which were easily separated by flash chromatography (Scheme 3). The minor Z-isomer could also provide novel stereoisomer analogues of RvD2 (1). Removal of the TIPS group with TBAF gave terminal alkyne 12. Alkyne 12 then underwent smooth hydrozirconation utilizing the procedure reported by Negishi [24] were ZrCp2HCl is generated in situ by reduction of ZrCp2Cl2 with DIBALH in THF. Iodinolysis of the zirconium species then gave the diene iodide 4 in good yield. Selectivity for this process was excellent with only a trace of the regioisomer formed.
![[1860-5397-9-310-i3]](/bjoc/content/inline/1860-5397-9-310-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Synthesis of dienyne 3
Our approach to the aldehyde 5 began with the production of the known bis-TES ether [9] produced by an alternative procedure (Scheme 4) in which the C7 stereochemistry was introduced via asymmetric dihydroxylation [25,26]. Thus, ester 13 [27] was treated with AD-mix-α in t-BuOH/H2O to give diol 14 in reasonable yield. The enantioselectivity and absolute configuration of the secondary alcohol was determined by conversion of diol into the bis-(S)-Mosher ester [28,29]. Integration of the 1H MMR spectrum indicated the e.r. was 93.7:6.3 and Mosher analysis (See Supporting Information File 2 for details) confirmed the stereochemistry of the new asymmetric center of the major enantiomer as S in accord with the predicted outcome [25]. Silylation gave bis-TES ether 15 and partial reduction of the alkyne using P2-Ni as catalyst [30] afforded the alkene 16. Desilylation of the primary TES group in 16 and concomitant oxidation to aldehyde 5 was achieved under Swern conditions as reported by Spur [31].
![[1860-5397-9-310-i4]](/bjoc/content/inline/1860-5397-9-310-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Deprotonation of salt 6 with LiHMDS followed by condensation with aldehyde 5 gave the E-enyne and the corresponding Z-isomer in a 4:1 ratio. The use of LiHMDS as base was critical for reasonable yields and stereoselectivity in this case. Global deprotection of 17 with TBAF gave the enyne 3 in good yield.
Total synthesis of resolvin D2 (1)
The completion of the synthesis of RvD2 (1) is shown in Scheme 5. Sonogashira coupling [32,33] between 3 and 4 was very efficient giving the alkyne 18 in good yield. Removal of the acetonide was effected by treatment with HCl in MeOH to give the known triol 19 [9]. The final steps to 1 were similar with those previously reported [5,9]. Thus, partial reduction of the triple bond using Zn(Cu/Ag) [34] to afforded RvD2 methyl ester 20 in 76% yield. A large excess of the Zn reagent was required to obtain a good conversion of 19 into 20. The 1H NMR spectrum (CDCl3 solvent) of RvD2 methyl ester (20) compared well to that reported [5]. Final ester hydrolysis and mild acid work-up then gave RvD2 (1).
![[1860-5397-9-310-i5]](/bjoc/content/inline/1860-5397-9-310-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Completion of the synthesis of RvD2 (1).
Scheme 5: Completion of the synthesis of RvD2 (1).
The synthetic RvD2 (1) had physical data identical to that reported [9,35] and we measured the specific rotation of this material for the first time ([α]D −17.5° (c 0.075, CH2Cl2)). In our hands, both RvD2 methyl ester (20) and RvD2 (1) itself were highly unstable, especially to acid. Prolonged standing in CDCl3 or CD3CN solution or exposure to light caused rapid decomposition and so NMR spectra were obtained quickly. We found that RvD2 methyl ester (20) was not very soluble in CD3CN so spectra were best run in CDCl3 that was filtered through basic alumina immediately prior to use. Spectra for RvD2 (1) were always measured for CD3CN. Even with short exposure to the solvent, we still observed degradation to unidentified compounds. Samples of RvD2 (1) can be stored in EtOH or frozen in DMSO solution but should be used immediately upon thawing. Alternatively, the triol 19 proved more stable than both RvD2 methyl ester (20) and RvD2 (1) and can be stored for longer periods prior to conversion to 1 which should be used rapidly for biological assessment to avoid degradation.
Conclusion
The total synthesis of RvD2 (1) has been completed using a common linchpin Wittig reaction. Using this approach, we were able to prepare sufficient quantities of this important inflammation resolving compound for further biological evaluation.
References
-
Serhan, C. N.; Hong, S.; Gronert, K.; Colgan, S. P.; Devchand, P. R.; Mirick, G.; Moussignac, R.-L. J. Exp. Med. 2002, 196, 1025–1037. doi:10.1084/jem.20020760
Return to citation in text: [1] [2] -
Serhan, C. N.; Petasis, N. A. Chem. Rev. 2011, 111, 5922–5943. doi:10.1021/cr100396c
Return to citation in text: [1] -
Hong, S.; Gronert, K.; Devchand, P. R.; Moussignac, R.-L.; Serhan, C. N. J. Biol. Chem. 2003, 278, 14677–14687. doi:10.1074/jbc.M300218200
Return to citation in text: [1] [2] -
Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N. A.; Serhan, C. N. J. Exp. Med. 2005, 201, 713–722. doi:10.1084/jem.20042031
Return to citation in text: [1] [2] -
Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Park, C.-K.; Xu, Z.-Z.; Liu, T.; Lü, N.; Serhan, C. N.; Ji, R.-R. J. Neurosci. 2011, 31, 18433–18438. doi:10.1523/JNEUROSCI.4192-11.2011
Return to citation in text: [1] -
Sun, Y.-P.; Oh, S. F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S. P.; Petasis, N. A.; Serhan, C. N. J. Biol. Chem. 2007, 282, 9323–9334. doi:10.1074/jbc.M609212200
Return to citation in text: [1] -
Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N. A.; Serhan, C. N. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1660–1665. doi:10.1073/pnas.0907342107
Return to citation in text: [1] -
Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2012, 53, 6990–6994. doi:10.1016/j.tetlet.2012.10.059
Return to citation in text: [1] -
Winkler, J. W.; Uddin, J.; Serhan, C. N.; Petasis, N. A. Org. Lett. 2013, 15, 1424–1427. doi:10.1021/ol400484u
Return to citation in text: [1] [2] -
Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2005, 46, 3623–3627. doi:10.1016/j.tetlet.2005.03.175
Return to citation in text: [1] -
Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2012, 53, 86–89. doi:10.1016/j.tetlet.2011.11.003
Return to citation in text: [1] -
Ogawa, N.; Kobayashi, Y. Tetrahedron Lett. 2009, 50, 6079–6082. doi:10.1016/j.tetlet.2009.08.061
Return to citation in text: [1] -
Allard, M.; Barnes, K.; Chen, X.; Cheung, Y.-Y.; Duffy, B.; Heap, C.; Inthavongsay, J.; Johnson, M.; Krishnamoorthy, R.; Manley, C.; Steffke, S.; Varughese, D.; Wang, R.; Wang, Y.; Schwartz, C. E. Tetrahedron Lett. 2011, 52, 2623–2626. doi:10.1016/j.tetlet.2011.03.035
Return to citation in text: [1] -
Ogawa, S.; Urabe, D.; Yokokura, Y.; Arai, H.; Arita, M.; Inoue, M. Org. Lett. 2009, 11, 3602–3605. doi:10.1021/ol901350g
Return to citation in text: [1] [2] -
Kosaki, Y.; Ogawa, N.; Kobayashi, Y. Tetrahedron Lett. 2010, 51, 1856–1859. doi:10.1016/j.tetlet.2010.01.109
Return to citation in text: [1] -
Urabe, D.; Todoroki, H.; Masuda, K.; Inoue, M. Tetrahedron 2012, 68, 3210–3219. doi:10.1016/j.tet.2012.02.045
Return to citation in text: [1] [2] -
Amin, R.; Chen, J.-X.; Cotterill, I. C.; Emrich, D.; Ganley, D.; Khmelnitsky, Y. L.; McLaws, M. D.; Michels, P. C.; Schwartz, C. E.; Thomas, D.; Yan, J.; Yang, Q. Org. Process Res. Dev. 2013, 17, 915–920. doi:10.1021/op4000384
Return to citation in text: [1] -
Current (Oct 2013) Australian price for 100 µg of RvD2 = $1,148 AUD. http://www.caymanchem.com/catalog/10007279.
Return to citation in text: [1] -
Marshall, J. A.; Salovich, J. M.; Shearer, B. G. J. Org. Chem. 1990, 55, 2398–2403. doi:10.1021/jo00295a030
Return to citation in text: [1] [2] -
Feutrill, J. T.; Lilly, M. J.; White, J. M.; Rizzacasa, M. A. Tetrahedron 2008, 64, 4880–4895. doi:10.1016/j.tet.2008.01.139
Return to citation in text: [1] [2] -
Corey, E. J.; Marfat, A.; Munroe, J.; Kim, K. S.; Hopkins, P. B.; Brion, F. Tetrahedron Lett. 1981, 22, 1077–1080. doi:10.1016/S0040-4039(01)90241-2
Return to citation in text: [1] -
Huang, Z.; Negishi, E.-i. Org. Lett. 2006, 8, 3675–3678. doi:10.1021/ol061202o
Return to citation in text: [1] -
Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispino, G. A.; Hartung, J.; Jeong, K. S.; Kwong, H. L.; Morikawa, K.; Wang, Z. M. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003
Return to citation in text: [1] [2] -
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009
Return to citation in text: [1] -
Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2012, 53, 4169–4172. doi:10.1016/j.tetlet.2012.05.143
Return to citation in text: [1] -
Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512–519. doi:10.1021/ja00783a034
Return to citation in text: [1] -
Hoye, T.; Jeffrey, C. S.; Shao, F. Nat. Protoc. 2007, 2, 2451–2458. doi:10.1038/nprot.2007.354
Return to citation in text: [1] -
Brown, C. A.; Ahuja, V. K. J. Chem. Soc., Chem. Commun. 1973, 553–554. doi:10.1039/C39730000553
Return to citation in text: [1] -
Rodríguez, A.; Nomen, M.; Spur, B. W.; Godfroid, J. J. Tetrahedron Lett. 1999, 40, 5161–5164. doi:10.1016/S0040-4039(99)00956-9
Return to citation in text: [1] -
Sonogashira, K. Coupling Reactions Between sp2 and sp Carbon Centers. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 3, pp 521–549. doi:10.1016/B978-0-08-052349-1.00071-8
Return to citation in text: [1] -
Nicolaou, K. C.; Veale, C. A.; Webber, S. E.; Katerinopoulos, H. J. Am. Chem. Soc. 1985, 107, 7515–7518. doi:10.1021/ja00311a048
Return to citation in text: [1] -
Boland, W.; Schroer, N.; Sieler, C.; Feigel, M. Helv. Chim. Acta 1987, 70, 1025–1040. doi:10.1002/hlca.19870700415
Return to citation in text: [1] -
One 13C NMR chemical shift (CD3CN solvent) was different to that reported. (34.6 ppm quoted as 31.2 ppm in [9]).
Return to citation in text: [1]
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 21. | Marshall, J. A.; Salovich, J. M.; Shearer, B. G. J. Org. Chem. 1990, 55, 2398–2403. doi:10.1021/jo00295a030 |
| 22. | Feutrill, J. T.; Lilly, M. J.; White, J. M.; Rizzacasa, M. A. Tetrahedron 2008, 64, 4880–4895. doi:10.1016/j.tet.2008.01.139 |
| 24. | Huang, Z.; Negishi, E.-i. Org. Lett. 2006, 8, 3675–3678. doi:10.1021/ol061202o |
| 1. | Serhan, C. N.; Hong, S.; Gronert, K.; Colgan, S. P.; Devchand, P. R.; Mirick, G.; Moussignac, R.-L. J. Exp. Med. 2002, 196, 1025–1037. doi:10.1084/jem.20020760 |
| 2. | Serhan, C. N.; Petasis, N. A. Chem. Rev. 2011, 111, 5922–5943. doi:10.1021/cr100396c |
| 3. | Hong, S.; Gronert, K.; Devchand, P. R.; Moussignac, R.-L.; Serhan, C. N. J. Biol. Chem. 2003, 278, 14677–14687. doi:10.1074/jbc.M300218200 |
| 5. | Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541 |
| 13. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2012, 53, 86–89. doi:10.1016/j.tetlet.2011.11.003 |
| 31. | Rodríguez, A.; Nomen, M.; Spur, B. W.; Godfroid, J. J. Tetrahedron Lett. 1999, 40, 5161–5164. doi:10.1016/S0040-4039(99)00956-9 |
| 1. | Serhan, C. N.; Hong, S.; Gronert, K.; Colgan, S. P.; Devchand, P. R.; Mirick, G.; Moussignac, R.-L. J. Exp. Med. 2002, 196, 1025–1037. doi:10.1084/jem.20020760 |
| 4. | Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N. A.; Serhan, C. N. J. Exp. Med. 2005, 201, 713–722. doi:10.1084/jem.20042031 |
| 14. | Ogawa, N.; Kobayashi, Y. Tetrahedron Lett. 2009, 50, 6079–6082. doi:10.1016/j.tetlet.2009.08.061 |
| 15. | Allard, M.; Barnes, K.; Chen, X.; Cheung, Y.-Y.; Duffy, B.; Heap, C.; Inthavongsay, J.; Johnson, M.; Krishnamoorthy, R.; Manley, C.; Steffke, S.; Varughese, D.; Wang, R.; Wang, Y.; Schwartz, C. E. Tetrahedron Lett. 2011, 52, 2623–2626. doi:10.1016/j.tetlet.2011.03.035 |
| 32. | Sonogashira, K. Coupling Reactions Between sp2 and sp Carbon Centers. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 3, pp 521–549. doi:10.1016/B978-0-08-052349-1.00071-8 |
| 33. | Nicolaou, K. C.; Veale, C. A.; Webber, S. E.; Katerinopoulos, H. J. Am. Chem. Soc. 1985, 107, 7515–7518. doi:10.1021/ja00311a048 |
| 4. | Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N. A.; Serhan, C. N. J. Exp. Med. 2005, 201, 713–722. doi:10.1084/jem.20042031 |
| 11. | Winkler, J. W.; Uddin, J.; Serhan, C. N.; Petasis, N. A. Org. Lett. 2013, 15, 1424–1427. doi:10.1021/ol400484u |
| 25. | Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispino, G. A.; Hartung, J.; Jeong, K. S.; Kwong, H. L.; Morikawa, K.; Wang, Z. M. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003 |
| 3. | Hong, S.; Gronert, K.; Devchand, P. R.; Moussignac, R.-L.; Serhan, C. N. J. Biol. Chem. 2003, 278, 14677–14687. doi:10.1074/jbc.M300218200 |
| 12. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2005, 46, 3623–3627. doi:10.1016/j.tetlet.2005.03.175 |
| 30. | Brown, C. A.; Ahuja, V. K. J. Chem. Soc., Chem. Commun. 1973, 553–554. doi:10.1039/C39730000553 |
| 8. | Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N. A.; Serhan, C. N. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1660–1665. doi:10.1073/pnas.0907342107 |
| 5. | Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541 |
| 27. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2012, 53, 4169–4172. doi:10.1016/j.tetlet.2012.05.143 |
| 7. | Sun, Y.-P.; Oh, S. F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S. P.; Petasis, N. A.; Serhan, C. N. J. Biol. Chem. 2007, 282, 9323–9334. doi:10.1074/jbc.M609212200 |
| 10. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2012, 53, 6990–6994. doi:10.1016/j.tetlet.2012.10.059 |
| 28. | Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512–519. doi:10.1021/ja00783a034 |
| 29. | Hoye, T.; Jeffrey, C. S.; Shao, F. Nat. Protoc. 2007, 2, 2451–2458. doi:10.1038/nprot.2007.354 |
| 6. | Park, C.-K.; Xu, Z.-Z.; Liu, T.; Lü, N.; Serhan, C. N.; Ji, R.-R. J. Neurosci. 2011, 31, 18433–18438. doi:10.1523/JNEUROSCI.4192-11.2011 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 5. | Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 25. | Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispino, G. A.; Hartung, J.; Jeong, K. S.; Kwong, H. L.; Morikawa, K.; Wang, Z. M. J. Org. Chem. 1992, 57, 2768–2771. doi:10.1021/jo00036a003 |
| 26. | Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483–2547. doi:10.1021/cr00032a009 |
| 11. | Winkler, J. W.; Uddin, J.; Serhan, C. N.; Petasis, N. A. Org. Lett. 2013, 15, 1424–1427. doi:10.1021/ol400484u |
| 16. | Ogawa, S.; Urabe, D.; Yokokura, Y.; Arai, H.; Arita, M.; Inoue, M. Org. Lett. 2009, 11, 3602–3605. doi:10.1021/ol901350g |
| 17. | Kosaki, Y.; Ogawa, N.; Kobayashi, Y. Tetrahedron Lett. 2010, 51, 1856–1859. doi:10.1016/j.tetlet.2010.01.109 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 18. | Urabe, D.; Todoroki, H.; Masuda, K.; Inoue, M. Tetrahedron 2012, 68, 3210–3219. doi:10.1016/j.tet.2012.02.045 |
| 5. | Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 34. | Boland, W.; Schroer, N.; Sieler, C.; Feigel, M. Helv. Chim. Acta 1987, 70, 1025–1040. doi:10.1002/hlca.19870700415 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 23. | Corey, E. J.; Marfat, A.; Munroe, J.; Kim, K. S.; Hopkins, P. B.; Brion, F. Tetrahedron Lett. 1981, 22, 1077–1080. doi:10.1016/S0040-4039(01)90241-2 |
| 5. | Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 21. | Marshall, J. A.; Salovich, J. M.; Shearer, B. G. J. Org. Chem. 1990, 55, 2398–2403. doi:10.1021/jo00295a030 |
| 22. | Feutrill, J. T.; Lilly, M. J.; White, J. M.; Rizzacasa, M. A. Tetrahedron 2008, 64, 4880–4895. doi:10.1016/j.tet.2008.01.139 |
| 19. | Amin, R.; Chen, J.-X.; Cotterill, I. C.; Emrich, D.; Ganley, D.; Khmelnitsky, Y. L.; McLaws, M. D.; Michels, P. C.; Schwartz, C. E.; Thomas, D.; Yan, J.; Yang, Q. Org. Process Res. Dev. 2013, 17, 915–920. doi:10.1021/op4000384 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 20. | Current (Oct 2013) Australian price for 100 µg of RvD2 = $1,148 AUD. http://www.caymanchem.com/catalog/10007279. |
| 16. | Ogawa, S.; Urabe, D.; Yokokura, Y.; Arai, H.; Arita, M.; Inoue, M. Org. Lett. 2009, 11, 3602–3605. doi:10.1021/ol901350g |
| 5. | Spite, M.; Norling, L. V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N. A.; Flower, R. J.; Perretti, M.; Serhan, C. N. Nature 2009, 461, 1287–1291. doi:10.1038/nature08541 |
| 18. | Urabe, D.; Todoroki, H.; Masuda, K.; Inoue, M. Tetrahedron 2012, 68, 3210–3219. doi:10.1016/j.tet.2012.02.045 |
| 9. | Rodríguez, A. R.; Spur, B. W. Tetrahedron Lett. 2004, 45, 8717–8720. doi:10.1016/j.tetlet.2004.09.129 |
| 35. | One 13C NMR chemical shift (CD3CN solvent) was different to that reported. (34.6 ppm quoted as 31.2 ppm in [9]). |
© 2013 Li et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)