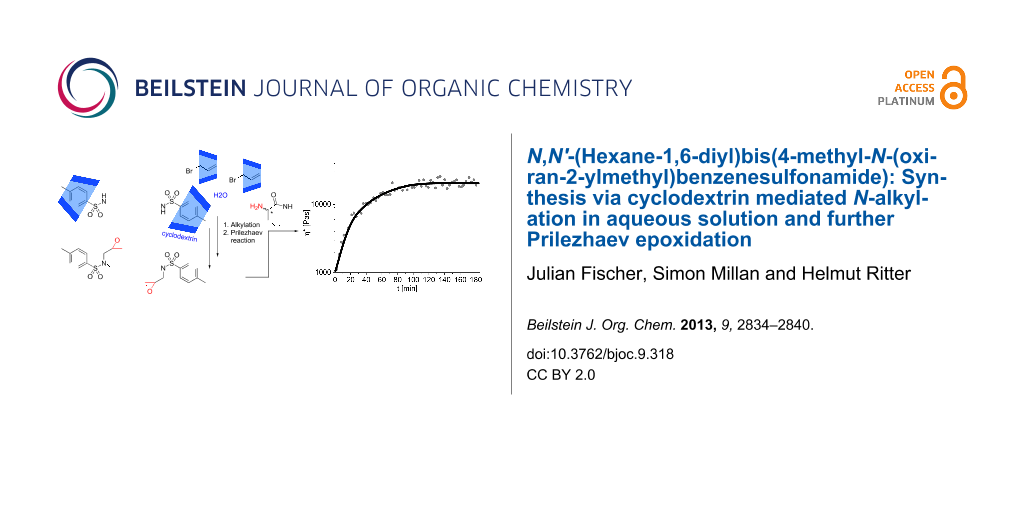
Abstract
N-alkylation of N,N'-(hexane-1,6-diyl)bis(4-methylbenzenesulfonamide) with allyl bromide and subsequent Prilezhaev reaction with m-chloroperbenzoic acid to give N,N'-(hexane-1,6-diyl)bis(4-methyl-N-(oxiran-2-ylmethyl)benzenesulfonamide) is described. This twofold alkylation was performed in aqueous solution, whereby α-, and randomly methylated β-cyclodextrin were used as adequate phase transfer catalysts and the cyclodextrin–guest complexes were characterized by 1H NMR and 2D NMR ROESY spectroscopy. Finally, the curing properties of the diepoxide with lysine-based α-amino-ε-caprolactam were analyzed by rheological measurements.

Graphical Abstract
Introduction
Various diepoxides easily react with amines or diamines to form cross-linked, cyclic or linear addition-polymers, which are implemented in construction, electronic, aerospace, medical and dental industries [1,2]. Hereby, bisphenol A diglycidyl ether (BADGE) is often used [3,4]. However, non-bisphenol A based diepoxides are subject to intensive research [5-9]. The industrial synthesis of BADGE and other commercially available diepoxides proceeds mainly by using epichlorohydrin, whereby usually several side products and oligomers are formed in significant concentrations [10-12]. An alternative route is a two-step reaction via N-allylation and further Prilezhaev epoxidation with peroxides [13-15]. The solubility of hydrophobic reactants in water can be increased significantly by cyclodextrins (CD) and thereby the use of organic solvents can be reduced [16-18]. To our best knowledge, CD mediated N-alkylation of sulfonamides is not yet described. Generally, only a few examples are known in literature about CD assisted alkylation of amines in aqueous solution [19-22]. Hence, in this work, we wish to present our direct and CD mediated method to alternative sulfonamide based diepoxides, focusing on one characteristic example. Furthermore, the polymerization of these types of diepoxides was investigated with lysine-based α-amino-ε-caprolactam through rheological measurements.
Results and Discussion
N,N'-(Hexane-1,6-diyl)bis(4-methylbenzenesulfonamide) (3), as precursor for the subsequent N-alkylation and further Prilezhaev epoxidation, was synthesized easily from p-toluenesulfonyl chloride (1) and hexamethylenediamine (2) [23]. The resulting crystalline sulfonamide was first described in 1927 prepared from 1,6-dibromohexane and p-toluenesulfonamide [24]. The subsequent two-fold N-alkylation of 3 with allyl bromide (4) was conducted in N,N-dimethylformamide as solvent, as well as in aqueous solution via CD-complexation (Scheme 1).
![[1860-5397-9-318-i1]](/bjoc/content/inline/1860-5397-9-318-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of sulfonamide 3, N-alkylation of 3 in organic solution and of CD-complex (3β) in aqueous phase to obtain 5 and the subsequent epoxidation with m-chloroperbenzoic acid (mCPBA) to yield product 6.
Scheme 1: Synthesis of sulfonamide 3, N-alkylation of 3 in organic solution and of CD-complex (3β) in aqueous...
Unmodified 3 is neither in a neutral nor in a basic milieu significantly soluble. However, by complexation of 3 with two equivalents of randomly methylated β-cyclodextrin (β-CD) to give 3β, water solubility could be increased distinctively. Characterization of the inclusion complex of β-CD with 3 in D2O solution was conducted using 2D NMR ROESY spectroscopy (Figure 1).
![[1860-5397-9-318-1]](/bjoc/content/figures/1860-5397-9-318-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 2D NMR ROESY spectrum of the complex of 3 with β-CD in D2O, displaying the interaction of the tosyl protons with the β-CD protons.
Figure 1: 2D NMR ROESY spectrum of the complex of 3 with β-CD in D2O, displaying the interaction of the tosyl...
Thereby, proton signals at 7.0 and 7.4 ppm can be assigned to the aromatic protons of 3. Furthermore, the protons of its p-methyl moiety demonstrate a singlet signal at 2.1 ppm. As illustrated in Figure 1, interaction of these protons with the inner protons of β-CD in the range of 3.3 to 3.8 ppm is visible. This strongly indicates a self-agglomeration of the tosyl groups of 3 with β-CD. Further confirmation of these findings is a perceivable downfield shifting of the proton signals of 3β compared to 3 suspended in D2O, since they are magnetically shielded by the β-CD cavity.
Additionally, the water solubility of 4 is limited as well (3.83 g L−1) [25]. Since the stability of CD–guest complexes often depends on the size of the guest molecule relative to the CD cavity, stability constants of unbranched alkyl chains or vinyl groups are the highest with α-CD (six glucose units) and for tosyl groups with β-CD (seven glucose units), respectively [18]. Therefore, the water solubility of 4 was increased by addition of α-CD (4α). 2D NMR ROESY spectroscopy was also performed for 4α. As expected, the protons of the allyl-group at 5.9 ppm, 5.1 ppm and 4.0 ppm correlate with the α-CD protons in the range of 3.9 to 3.5 ppm (Figure S1, Supporting Information File 1).
Two-fold N-alkylation of β-CD-complexed N,N'-(hexane-1,6-diyl)bis(4-methylbenzenesulfonamide) (3β) with 4α in aqueous solution gave N,N'-(hexane-1,6-diyl)bis(N-allyl-4-methylbenzenesulfonamide) (5), which could easily be precipitated and separated from the CD-complex by heating, since decomplexation occurred over 70 °C. The remaining aqueous CD-solution can be reused in principle, since in an alkaline milieu CD-rings are stable. A comparison of the 1H NMR spectra of 5 received in organic and in aqueous solution, respectively is given in Supporting Information File 1 (Figure S2) to show no significant differences. Also, CD signals in the range of 3.0 to 4.0 ppm are not observable. That indicates complete CD-decomplexation of 5. Subsequently, 5 was epoxidized in a Prilezhaev reaction with m-chloroperbenzoic acid (mCPBA) in methylene chloride to obtain N,N'-(hexane-1,6-diyl)bis(4-methyl-N-(oxiran-2-ylmethyl)benzenesulfonamide) (6). The proceeding reaction was monitored by means of 1H NMR spectroscopy, since the allyl protons of 5 in the range of 5.0 to 5.7 ppm vanish on conversion (Figure S3, Supporting Information File 1). To illustrate the curing properties of the synthesized diepoxide, 6 was reacted in a ring-opening polymerization with the primary amine α-amino-ε-caprolactam (8). 8 was synthesized by cyclization of lysine (7) (Scheme 2). Hence, an increase of the reactivity of the primary amino group towards the epoxide function compared to the amino groups in native lysine was expected [26-28].
![[1860-5397-9-318-i2]](/bjoc/content/inline/1860-5397-9-318-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cyclization of L-(+)-lysine monohydrochloride to give racemate 8.
Scheme 2: Cyclization of L-(+)-lysine monohydrochloride to give racemate 8.
As illustrated in Figure 2A, oscillatory measurements display an initial high viscosity for a mixture of 6 and 8, since its complex viscosity (η*), calculated from storage modulus G’ and loss modulus G’’, exhibits low four-digit values at 50 °C. However, η* increases further on time until the sol–gel-transition (gelpoint), where G’ is equal to G’’, is reached after about 40 to 45 min (Figure 2B). Generally, by passing this point the mixture is no longer capable of flowing. Shortly after reaching the gelpoint, η* is not significantly rising further, which is a sign for completed conversion. The resulting poly-adduct 9 exhibits a glass transition temperature (Tg) of around 49 °C. Equivalent measurements of standard BADGE demonstrated a gelpoint after about 99 min and a Tg of about 74 °C (Figure S4, Supporting Information File 1). Thus, the curing properties for a mixture of 6 with 8 are relatively comparable to a mixture of BADGE with 8, whereby the observed differences can be caused by different reactivity of the oxirane moieties or unequal solubility of the respective diepoxide with 8.
![[1860-5397-9-318-2]](/bjoc/content/figures/1860-5397-9-318-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Oscillatory rheological measurements of an equimolar mixture of 6 and 8 at 50 °C. Illustrated is the complex viscosity (A), as well as G’ and G’’ (B) in dependence of time. The polymer 9 is displayed as an idealized structure (R = H or polymer network; crosslinking via addition of epoxide moieties to the hydroxy functions is likely [2]).
Figure 2: Oscillatory rheological measurements of an equimolar mixture of 6 and 8 at 50 °C. Illustrated is th...
By means of IR spectroscopy, ring opening polymerization of epoxides with amines can be monitored. The spectrum of 6 exhibits weak bands at 1253 and 895 cm−1, which can be assigned to the C–O-stretching vibration and the symmetric ring deformation vibration, respectively, of its epoxide groups. On curing at 50 °C, a broad band between 3100 and 3600 cm−1 appears which is caused by hydrogen bonded hydroxy stretching vibrations originating from epoxide ring opening. The epoxide bands seem to vanish after curing, which is a sign for high conversion. However, due to overlaps of broader bands in adjacent areas, no clear statement can be made in this regard (Figure S5, Supporting Information File 1). Based on this work, future research could focus on conducting the entire synthesis of 6 CD mediated in aqueous solution. Also, the presented pathway for alkylation in aqueous media could be transferred to the solvent saving synthesis of further alkylated products.
Conclusion
An alternative route for the synthesis of N,N'-(hexane-1,6-diyl)bis(4-methyl-N-(oxiran-2-ylmethyl)benzenesulfonamide) (6) via N-alkylation of N,N'-(hexane-1,6-diyl)bis(4-methylbenzenesulfonamide) (3) and further oxidation was described. For the first time, the CD-mediated N-alkylation of a sulfonamide was conducted, whereby water solubility of 3 and 4 were increased by adding β-CD and α-CD, respectively. By applying oscillatory rheological measurements, a mixture of 6 and 8 reached the gelpoint after about 40 to 45 min at 50 °C. Thus, an environmentally more sustainable route for the synthesis of a bisphenol A free diepoxide was presented, which appears to be a suitable substitute for technically employed bisphenol A diglycidyl ether.
Experimental
All reactants were commercially available and unless otherwise stated used without further purification. All solvents used were of analytical purity or freshly distilled. β-CD and α-CD were obtained from Wacker Chemie GmbH and were used after being dried with a vacuum oil pump over P4O10. p-Toluenesulfonyl chloride (98%) was obtained from Alfa Aesar, allyl bromide (99%), 3-chloroperoxybenzoic acid (70–75%), 1,6-hexanediamine (99.5+%) and L-(+)-lysine monohydrochloride (99+%) were purchased from Acros Organics, bisphenol A diglycidyl ether and deuterium oxide (99.9%) were provided from Sigma Aldrich, chloroform-d (99.80%) was obtained from Euriso-Top and dimethyl sulfoxide-d6 (99.9%) was purchased from Deutero. 1H NMR, 13C NMR and 2D ROESY spectra were recorded on a Bruker Avance DRX 300 and a Bruker Avance III – 600 by using deuterium oxide, dimethyl sulfoxide-d6 or chloroform-d as solvents. The chemical shifts (δ) are given in parts per million (ppm) using the solvent peak as an internal standard. FTIR spectra were recorded on a Nicolet 6700 FTIR spectrometer equipped with an ATR unit. Oscillatory rheological measurements were performed on a Haake Mars II rheometer by ThermoFisher Scientific. For this purpose a plate-plate construction (MP35, PP35Ti) was used. The temperature was set to 50 °C and was determined in the measuring plate with an accuracy of ±0.1 °C. The Sol–gel-transmission was determined by the first intersection of G’ and G’’. Graphs were plotted using reduced data, calculated by the median of three following values. The glass transition temperature (Tg) was determined using a Mettler Toledo DSC 822e equipped with a sample robot TSO801RO. For calibration, standard tin, indium, and zinc samples were used. Heating and cooling curves were determined between −30 and 130 °C at a heating rate of 15 °C/min. The Tg value was taken from the inflection point of the second heating curve. Electrospray ionization mass spectrometry (ESIMS) was conducted on a Bruker maXis 4G mass spectrometer. Melting points were obtained using a Büchi Melting Point B-545 apparatus at a heating rate of 5 °C/min.
Synthesis of 5 in organic solution: 7.0 mmol of 3 were dissolved in 25 mL dried N,N-dimethylformamide and the solution was cooled to 0 °C in an ice bath. The apparatus was set under constant nitrogen-flow and 17.2 mmol of sodium hydride were added. Afterwards, the suspension was stirred for 2 h and subsequently, 35 mmol of allyl bromide (4), which was filtrated through neutral aluminium oxide, was added dropwise. The resulting mixture was stirred at 50 °C for 18 h and after cooling to room temperature it was diluted with 40 mL of saturated solution of ammonium chloride. The aqueous phase was washed threefold with 40 mL of ethyl acetate, the organic phases were combined and the solvent was evaporated under reduced pressure. After drying, 5.3 mmol (76% yield, not optimized) of brown slurry were received. The raw product was purified by column chromatography (n-hexane/ethyl acetate 2:1) to give 3.98 mmol (57% yield, not optimized) of 5. 1H NMR (300 MHz, DMSO-d6, δ) 7.68 (d, J = 8.3 Hz, 4H, Ar-H), 7.40 (d, J = 7.9 Hz, 4H, Ar-H), 5.60 (ddt, J = 6.3, 10.1, 17.1 Hz, 2H, H2C=CH-CH2-), 5.19 (dd, J = 17.0, 1.6 Hz, 2H, CH2, trans), 5.11 (dd, J = 10.1, 1.6 Hz, 2H, CH2, cis), 3.73 (d, 4H, J = 6.4 Hz, allyl-CH2-), 2.99 (t, J = 7.4 Hz, 4H, N-CH2-), 2.39 (s, 6H, Ar-CH3), 1.38 (m, 4H, -CH2-), 1.14 (m, 4H, -CH2-); 13C NMR (75 MHz, DMSO-d6, δ) 143.0 (2C, Ar(C)-CH3), 136.6 (2C, RO2S-(C)Ar), 133.5 (2C, R-CH=CH2), 129.8 (4C, Ar(C)), 126.9 (4C, Ar(C)), 118.5 (2C, RHC=CH2), 50.1 (2C, N-CH2-CHR), 47.0 (2C, N-CH2-CH2R), 27.5 (2C, -CH2-), 25.5 (2C, -CH2-), 20.9 (2C, -CH3); IR (diamond) ν (cm−1): 2951, 2916, 2855 (m, -CH2-, Ar-CH3), 1652 (w, C=C), 1597 (m, Ar), 1336, 1155 (v, R-SO2-NR2), 1090 (m, Ar-S-), 965 (v, -CH2-), 934 (m, R-SO2-NR2), 816 (s, Ar-H (neighbouring)); ESIMS m/z: 505.4 [M + H]+, 527 [M + Na]+; mp 70 °C.
Synthesis of 5 CD mediated in aqueous solution: 6.8 g of randomly methylated β-CD and 25 mmol of sodium hydroxide were dissolved in 15 mL double distilled water and 2.4 mmol of 3 were added. The initial suspension was stirred at 45 °C for some minutes until a homogenous solution was achieved. Simultaneously, a homogenous solution of 1.15 mmol of 4, 13.8 mmol of sodium hydroxide and 1.6 mmol of α-CD in 10 mL of double distilled water was prepared. Subsequently, the solution of 4α was added dropwise to the solution of 3β. The combined solutions were stirred at 50 °C and over the next 15 h further 8 mmol of 4 were added successively. After two days, the solution was heated to 70 °C for 30 min, filtrated and the precipitate dried in vacuo. 0.9 mmol (38% yield, not optimized) of a colorless powder of 5 was obtained. Spectral data of 5 synthesized in aqueous solution are given in the Supporting Information File 1.
Synthesis of 6: 3.0 mmol of 5 were dissolved in 15 mL of methylene chloride. Subsequently, 12.6 mmol of mCPBA dissolved in 20 mL methylene chloride were added. After two days of stirring at room temperature the reaction batch was treated with further 8.7 mmol of mCPBA and stirred for 4 days. Afterwards, the suspension was filtrated, the filtrate diluted with 20 mL of methylene chloride and washed three times with an aqueous solution of 10% sodium bisulfite. Then, the organic phase was treated twice with 50 mL each of sodium hydrogen carbonate solution and stirred for 1.5 h. Subsequently, the organic phase was dried over magnesium sulfate and the solvent removed under reduced pressure to obtain 1.7 mmol (57% yield, not optimized) of 6. 1H NMR (300 MHz, CDCl3, δ): 7.67 (d, J = 8.3 Hz, 4H, Ar-H), 7.28 (d, J = 7.9 Hz, 4H, Ar-H), 3.59 (dd, J = 3.4, 15.2 Hz, 2H, N-CH2-CH-, trans), 3.15 (m, 4H, N-CH2-CH2-), 3.05 (m, 2H, -CH-), 2.87 (dd, J = 6.5, 15.2 Hz, 2H, N-CH2-CH-, cis), 2.74 (dd, J = 5.3, 4.2 Hz, 2H, O–CH2, cis), 2.49 (dd, J = 2.6, 4.3 Hz, 2H, O–CH2, trans), 2.40 (s, 6H, Ar-CH3), 1.55 (m, 4H, -CH2-), 1.27 (m, 4H, -CH2-); 13C NMR (75 MHz, CDCl3, δ) 143.6 (2C, Ar(C)-CH3), 136.8 (2C, RO2S-(C)Ar), 130.0 (4C, Ar(C)), 127.3 (4C, Ar(C)), 51.2 (2C, epoxy), 51.0 (2C, N-CH2-CH2R), 49.4 (2C, N-CH2-epoxy), 45.4 (2C, epoxy), 28.5 (2C, -CH2-), 26.3 (2C, -CH2-), 21.6 (2C, -CH3); IR (diamond) ν (cm−1): 2997, 2927, 2862 (m, -CH2-, Ar-CH3), 1597 (m, Ar), 1435 (m, -CH2-), 1335 (v, R-SO2-NR2), 1253 (w, -C-O-C-), 1154 (v, R-SO2-NR2), 1090 (m, Ar-S-), 895 (w, -C-O-C-), 839 (m, C-C (ring)), 816 (s, Ar-H (neighbouring)); ESIMS m/z: 537.1 [M + H]+, 559.1 [M + Na]+.
Synthesis of 3 and 8: Sulfonamide 3 was obtained in a modified synthesis similar to that reported in [23]. The synthesis of 8 was performed according to [29].
Supporting Information
Experimental procedures and spectral data for the synthesis of 3 and 8 are given in the Supporting Information.
| Supporting Information File 1: Additional spectra and experimental data. | ||
| Format: PDF | Size: 620.8 KB | Download |
References
-
Klee, J. E.; Flammersheim, H.-J. Macromol. Chem. Phys. 2002, 203, 100–108. doi:10.1002/1521-3935(20020101)203:1<100::AID-MACP100>3.0.CO;2-J
Return to citation in text: [1] -
Klee, J.; Hörhold, H.-H. Epoxide-amine addition polymers, linear. In Polymeric Materials Encyclopedia; Salamone, J. C., Ed.; CRC Press: Boca Raton, Florida, USA, 1996; Vol. 3, D-E, pp 2182–2192.
Return to citation in text: [1] [2] -
Wang, L.; Wu, Y.; Zhang, W.; Kannan, K. Environ. Sci. Technol. 2012, 46, 12968–12976. doi:10.1021/es304050f
Return to citation in text: [1] -
Resende, L. M.; Rached-Junior, F. J. A.; Versiani, M. A.; Souza-Gabriel, A. E.; Miranda, C. E. S.; Silva-Sousa, Y. T. C.; Sousa Neto, M. D. Int. Endodont. J. 2009, 42, 785–793. doi:10.1111/j.1365-2591.2009.01584.x
Return to citation in text: [1] -
Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.-P. Prog. Polym. Sci. 2010, 35, 578–622. doi:10.1016/j.progpolymsci.2009.10.001
Return to citation in text: [1] -
Chiu, Y.-C.; Chou, I. C.; Tseng, W.-C.; Ma, C.-C. M. Polym. Degrad. Stab. 2008, 93, 668–676. doi:10.1016/j.polymdegradstab.2007.12.014
Return to citation in text: [1] -
Shikha, D.; Kamani, P. K.; Shukla, M. C. Prog. Org. Coat. 2003, 47, 87–94. doi:10.1016/S0300-9440(02)00159-5
Return to citation in text: [1] -
Tao, Z.; Yang, S.; Chen, J.; Fan, L. Eur. Polym. J. 2007, 43, 1470–1479. doi:10.1016/j.eurpolymj.2007.01.039
Return to citation in text: [1] -
Pews, R. G. Diepoxide derivatives of N,N*-disubstituted disulfonamides. U.S. Patent 0128978 A1, June 15, 2006.
Return to citation in text: [1] -
Kolman, A.; Chovanec, M.; Osterman-Golkar, S. Mutat. Res. 2002, 512, 173–194. doi:10.1016/S1383-5742(02)00067-4
Return to citation in text: [1] -
Braun, D.; Lee, D. W. Angew. Makromol. Chem. 1976, 51, 11–24. doi:10.1002/apmc.1976.050510102
Return to citation in text: [1] -
Braun, D.; Lee, D. W. Angew. Makromol. Chem. 1977, 57, 111–122. doi:10.1002/apmc.1977.050570109
Return to citation in text: [1] -
Prileschajew, N. Ber. Dtsch. Chem. Ges. 1909, 42, 4811–4815. doi:10.1002/cber.190904204100
Return to citation in text: [1] -
Woods, K. W.; Beak, P. J. Am. Chem. Soc. 1991, 113, 6281–6283. doi:10.1021/ja00016a061
Return to citation in text: [1] -
Mimoun, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 734–750. doi:10.1002/anie.198207341
Return to citation in text: [1] -
Ritter, H.; Tabatabai, M. Prog. Polym. Sci. 2002, 27, 1713–1720. doi:10.1016/S0079-6700(02)00022-9
Return to citation in text: [1] -
Alupei, V.; Alupei, I. C.; Ritter, H. Macromol. Rapid Commun. 2005, 26, 40–45. doi:10.1002/marc.200400442
Return to citation in text: [1] -
Wenz, G. Angew. Chem., Int. Ed. Engl. 1994, 33, 803–822. doi:10.1002/anie.199408031
Return to citation in text: [1] [2] -
Surendra, K.; Krishnaveni, N. S.; Sridhar, R.; Rao, K. R. Tetrahedron Lett. 2006, 47, 2125–2127. doi:10.1016/j.tetlet.2006.01.124
Return to citation in text: [1] -
Zhang, Q.; Cheng, G.; Huang, Y.-Z.; Qu, G.-R.; Niu, H.-Y.; Guo, H.-M. Tetrahedron 2012, 68, 7822–7826. doi:10.1016/j.tet.2012.07.025
Return to citation in text: [1] -
Yamaguchi, I.; Osakada, K.; Yamamoto, T. Macromolecules 1997, 30, 4288–4294. doi:10.1021/ma970051p
Return to citation in text: [1] -
Li, W.; Zhang, W.; Ma, X.; Wang, P.; Du, M. Appl. Catal., A: Gen. 2012, 419–420, 210–214. doi:10.1016/j.apcata.2012.01.030
Return to citation in text: [1] -
Stetter, H.; Roos, E.-E. Chem. Ber. 1954, 87, 566–571. doi:10.1002/cber.19540870421
Return to citation in text: [1] [2] -
Müller, A.; Sauerwald, A. Monatsh. Chem. 1927, 48, 727–732.
Return to citation in text: [1] -
Tewari, Y. B.; Miller, M. M.; Wasik, S. P.; Martire, D. E. J. Chem. Eng. Data 1982, 27, 451–454. doi:10.1021/je00030a025
Return to citation in text: [1] -
Philippe, C.; Milcent, T.; Crousse, B.; Bonnet-Delpon, D. Org. Biomol. Chem. 2009, 7, 2026–2028. doi:10.1039/b902081k
Return to citation in text: [1] -
Brotzel, F.; Chu, Y. C.; Mayr, H. J. Org. Chem. 2007, 72, 3679–3688. doi:10.1021/jo062586z
Return to citation in text: [1] -
Brotzel, F.; Mayr, H. Org. Biomol. Chem. 2007, 5, 3814–3820. doi:10.1039/b713778h
Return to citation in text: [1] -
Frost, J. W. Synthesis of Caprolactam from Lysine. WO Patent 2005/123669 A1, Dec 29, 2005.
Return to citation in text: [1]
| 1. | Klee, J. E.; Flammersheim, H.-J. Macromol. Chem. Phys. 2002, 203, 100–108. doi:10.1002/1521-3935(20020101)203:1<100::AID-MACP100>3.0.CO;2-J |
| 2. | Klee, J.; Hörhold, H.-H. Epoxide-amine addition polymers, linear. In Polymeric Materials Encyclopedia; Salamone, J. C., Ed.; CRC Press: Boca Raton, Florida, USA, 1996; Vol. 3, D-E, pp 2182–2192. |
| 13. | Prileschajew, N. Ber. Dtsch. Chem. Ges. 1909, 42, 4811–4815. doi:10.1002/cber.190904204100 |
| 14. | Woods, K. W.; Beak, P. J. Am. Chem. Soc. 1991, 113, 6281–6283. doi:10.1021/ja00016a061 |
| 15. | Mimoun, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 734–750. doi:10.1002/anie.198207341 |
| 29. | Frost, J. W. Synthesis of Caprolactam from Lysine. WO Patent 2005/123669 A1, Dec 29, 2005. |
| 10. | Kolman, A.; Chovanec, M.; Osterman-Golkar, S. Mutat. Res. 2002, 512, 173–194. doi:10.1016/S1383-5742(02)00067-4 |
| 11. | Braun, D.; Lee, D. W. Angew. Makromol. Chem. 1976, 51, 11–24. doi:10.1002/apmc.1976.050510102 |
| 12. | Braun, D.; Lee, D. W. Angew. Makromol. Chem. 1977, 57, 111–122. doi:10.1002/apmc.1977.050570109 |
| 5. | Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.-P. Prog. Polym. Sci. 2010, 35, 578–622. doi:10.1016/j.progpolymsci.2009.10.001 |
| 6. | Chiu, Y.-C.; Chou, I. C.; Tseng, W.-C.; Ma, C.-C. M. Polym. Degrad. Stab. 2008, 93, 668–676. doi:10.1016/j.polymdegradstab.2007.12.014 |
| 7. | Shikha, D.; Kamani, P. K.; Shukla, M. C. Prog. Org. Coat. 2003, 47, 87–94. doi:10.1016/S0300-9440(02)00159-5 |
| 8. | Tao, Z.; Yang, S.; Chen, J.; Fan, L. Eur. Polym. J. 2007, 43, 1470–1479. doi:10.1016/j.eurpolymj.2007.01.039 |
| 9. | Pews, R. G. Diepoxide derivatives of N,N*-disubstituted disulfonamides. U.S. Patent 0128978 A1, June 15, 2006. |
| 2. | Klee, J.; Hörhold, H.-H. Epoxide-amine addition polymers, linear. In Polymeric Materials Encyclopedia; Salamone, J. C., Ed.; CRC Press: Boca Raton, Florida, USA, 1996; Vol. 3, D-E, pp 2182–2192. |
| 3. | Wang, L.; Wu, Y.; Zhang, W.; Kannan, K. Environ. Sci. Technol. 2012, 46, 12968–12976. doi:10.1021/es304050f |
| 4. | Resende, L. M.; Rached-Junior, F. J. A.; Versiani, M. A.; Souza-Gabriel, A. E.; Miranda, C. E. S.; Silva-Sousa, Y. T. C.; Sousa Neto, M. D. Int. Endodont. J. 2009, 42, 785–793. doi:10.1111/j.1365-2591.2009.01584.x |
| 23. | Stetter, H.; Roos, E.-E. Chem. Ber. 1954, 87, 566–571. doi:10.1002/cber.19540870421 |
| 18. | Wenz, G. Angew. Chem., Int. Ed. Engl. 1994, 33, 803–822. doi:10.1002/anie.199408031 |
| 23. | Stetter, H.; Roos, E.-E. Chem. Ber. 1954, 87, 566–571. doi:10.1002/cber.19540870421 |
| 26. | Philippe, C.; Milcent, T.; Crousse, B.; Bonnet-Delpon, D. Org. Biomol. Chem. 2009, 7, 2026–2028. doi:10.1039/b902081k |
| 27. | Brotzel, F.; Chu, Y. C.; Mayr, H. J. Org. Chem. 2007, 72, 3679–3688. doi:10.1021/jo062586z |
| 28. | Brotzel, F.; Mayr, H. Org. Biomol. Chem. 2007, 5, 3814–3820. doi:10.1039/b713778h |
| 19. | Surendra, K.; Krishnaveni, N. S.; Sridhar, R.; Rao, K. R. Tetrahedron Lett. 2006, 47, 2125–2127. doi:10.1016/j.tetlet.2006.01.124 |
| 20. | Zhang, Q.; Cheng, G.; Huang, Y.-Z.; Qu, G.-R.; Niu, H.-Y.; Guo, H.-M. Tetrahedron 2012, 68, 7822–7826. doi:10.1016/j.tet.2012.07.025 |
| 21. | Yamaguchi, I.; Osakada, K.; Yamamoto, T. Macromolecules 1997, 30, 4288–4294. doi:10.1021/ma970051p |
| 22. | Li, W.; Zhang, W.; Ma, X.; Wang, P.; Du, M. Appl. Catal., A: Gen. 2012, 419–420, 210–214. doi:10.1016/j.apcata.2012.01.030 |
| 16. | Ritter, H.; Tabatabai, M. Prog. Polym. Sci. 2002, 27, 1713–1720. doi:10.1016/S0079-6700(02)00022-9 |
| 17. | Alupei, V.; Alupei, I. C.; Ritter, H. Macromol. Rapid Commun. 2005, 26, 40–45. doi:10.1002/marc.200400442 |
| 18. | Wenz, G. Angew. Chem., Int. Ed. Engl. 1994, 33, 803–822. doi:10.1002/anie.199408031 |
| 25. | Tewari, Y. B.; Miller, M. M.; Wasik, S. P.; Martire, D. E. J. Chem. Eng. Data 1982, 27, 451–454. doi:10.1021/je00030a025 |
© 2013 Fischer et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)