Abstract
An iron(II)-mediated aminohalogenation of a cyclopentenyl N-tosyloxycarbamate provided new access to the key intermediate for the synthesis of (−)-agelastatin A (AA, 1), a potent antiproliferative alkaloid. The present synthetic endeavour offered an insight into the mechanism underlying the iron(II)-mediated aminohalogenation of N-tosyloxycarbamate, in which the radical properties of the N–iron intermediates in the redox states were operative.

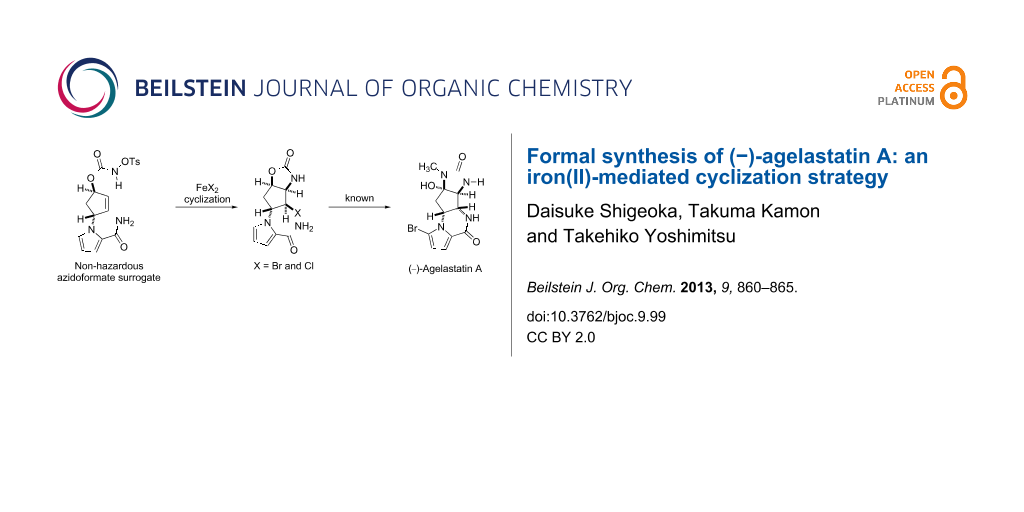
Graphical Abstract
Introduction
Marine organisms often produce bioactive substances that potentially serve as attractive resources for drug discovery. (−)-Agelastatin A (AA, 1), a cytotoxic alkaloid isolated from marine sponges Agelas dendromorpha and Cymbastela sp., is one such substance, which has drawn considerable attention due to its potential applicability in the development of anticancer agents [1-5]. The intriguing biological activity of 1 has stimulated interest in developing various chemical accesses to the natural product [6-25]. Our previous synthetic endeavours have established two approaches to 1, in which cyclopentenyl azidoformates 2 and 3 were utilized as the pivotal intermediates (Scheme 1).
![[1860-5397-9-99-i1]](/bjoc/content/inline/1860-5397-9-99-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Our first- [26] and second-generation [27] approaches to (−)-agelastatin A (1).
Scheme 1: Our first- [26] and second-generation [27] approaches to (−)-agelastatin A (1).
The first-generation strategy employed a stereoselective thermal aziridination of azidoformate 2 and a subsequent aziridine-opening reaction to establish the vicinal trans nitrogen motif 4 [26]. The second-generation strategy involved the radical aminobromination of azidoformate 3 followed by lactamization of the resultant bromide 5a to furnish a tetracyclic compound (structure not shown), which was transformed into the natural product [27]. In the present study, we disclose a new approach to the key intermediate for AA synthesis in which N-tosyloxycarbamate 8, a nonhazardous azidoformate surrogate, is transformed into aminohalogenated compounds 5a and 5b by FeBr2/Bu4NBr [28,29], FeCl2/Bu4NCl, or FeCl2/TMSCl [30-35] (Scheme 2). Moreover, a plausible mechanism of the present iron(II)-mediated aminohalogenation, which is inferred from the unique reactivity of N-tosyloxycarbamate 8 with the reagents, is discussed.
![[1860-5397-9-99-i2]](/bjoc/content/inline/1860-5397-9-99-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The present iron(II)-mediated aminohalogenation of N-tosyloxycarbamate 8 providing key intermediates 5a/5b for (−)-agelastatin A synthesis.
Scheme 2: The present iron(II)-mediated aminohalogenation of N-tosyloxycarbamate 8 providing key intermediate...
Results and Discussion
N-Tosyloxycarbamate 8 was prepared from alcohol 6, which was obtained by a previously reported protocol (Scheme 2) [26,27]. Alcohol 6 was first treated with CDI (N,N’-carbonyldiimidazole) and then with hydroxylamine hydrochloric acid salt to afford N-hydroxycarbamate 7 in 67% yield [36]. Thereafter, N-hydroxycarbamate 7 was reacted with TsCl and triethylamine in THF to furnish N-tosyloxycarbamate 8 in 92% yield. With this carbamate 8, we examined the iron(II)-mediated cyclization under various conditions (Table 1).
Table 1: Aminohalogenation of N-tosyloxycarbamate 8 by iron(II) catalysis.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-99-i6.svg?max-width=637&scale=1.0)
|
||
| entry | conditionsa | products (%) |
|---|---|---|
| 1 | FeBr2 (0.5 equiv), Bu4NBr (1.5 equiv), EtOH, rt, 1.75 h | 5a (13), 9 (39), 10 (30) |
| 2 | FeCl2 (0.5 equiv), Bu4NCl (1.2 equiv), EtOH, rt, 0.75 h | 5b (39), 9 (20), 10 (19) |
| 3 | FeBr2 (0.5 equiv), Bu4NBr (1.2 equiv), t-BuOH, rt, 0.5 h | 5a (38), 9 (22), 10 (19) |
| 4 | FeCl2 (0.5 equiv), Bu4NCl (1.2 equiv), t-BuOH, rt, 2.5 h | 5b (48), 9 (9), 10 (9) |
| 5 | FeBr2 (0.2 equiv), Bu4NBr (1.2 equiv), t-BuOH, rt, 3.3 h | 5a (25)b, 9 (16), 10 (5) |
| 6 | FeCl2 (0.2 equiv), Bu4NCl (1.2 equiv), t-BuOH, rt, 3.3 h | 5b (31)c, 9 (9), 10 (14) |
| 7 | FeCl2 (0.5 equiv), TMSCl (1.5 equiv), EtOH, 0 °C to rt, 16 h | 5b (29), 9 (12)d |
aAll reactions were conducted using 20 mg of substrate 8. b22% of 8 was recovered. c35% of 8 was recovered. dCompounds 11 (16%) and 12 (14%) were obtained.
The application of FeBr2 (0.5 equiv)/Bu4NBr (1.5 equiv) in EtOH effected cyclization, but the yield of 5a was poor due to the concomitant formation of carbamate 9 (39%) and enone 10 (30%) (Table 1, entry 1). This was in marked contrast to the observation that the same reagent system, i.e., FeBr2 (0.5 equiv)/Bu4NBr (1.2 equiv), allowed the efficient conversion of azidoformate 3 (2 g scale) in EtOH to afford 5a in 70% yield (Scheme 3). The distinct yields of the cyclized materials obtained from N-tosyloxycarbamate 8 and azidoformate 3 suggested the unique reactivity of each substrate towards the iron(II) halide (see below). An aminochlorination reagent system, i.e., FeCl2 (0.5 equiv)/Bu4NCl (1.2 equiv) in EtOH, in turn, furnished the corresponding chloride 5b in 39% yield as the major product (Table 1, entry 2). Our recent studies on the iron(II)-mediated aminobromination reactions of structurally simple N-tosyloxycarbamates with FeBr2/Bu4NBr revealed significant solvent effects on the product yields [28]. This was also the case in the present study: FeBr2 (0.5 equiv)/Bu4NBr (1.2 equiv) in t-BuOH successfully improved the yield of 5a relative to the reaction in EtOH (Table 1, entry 3). FeCl2 (0.5 equiv)/Bu4NCl (1.2 equiv) in t-BuOH culminated in the highest yield of 5b among the examined conditions (Table 1, entry 4) However, a reduction of FeX2 loading even in t-BuOH led to erosion of the yields of halides 5a and 5b with recovery of the substrate (Table 1, entries 5 and 6). With the FeCl2/TMSCl reagent system [30-35], chloride 5b was accessible from N-tosyloxycarbamate 8 in 29% yield, along with 9 in 12% yield (Table 1, entry 7). In this particular case, cyclopentanone derivative 11 (16%) and diethyl ketal 12 (14%) were produced as well (Figure 1). An additional experiment to elucidate the origin of their formation provided evidence that these byproducts were generated by the intramolecular cyclization of enone 10 with TMSCl in EtOH, suggesting that the FeCl2/TMSCl system also gave enone 10 in ca. 30% yield [37].
![[1860-5397-9-99-i3]](/bjoc/content/inline/1860-5397-9-99-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Aminohalogenation of azidoformate 3 (2 g scale) under FeBr2/Bu4NBr conditions.
Scheme 3: Aminohalogenation of azidoformate 3 (2 g scale) under FeBr2/Bu4NBr conditions.
![[1860-5397-9-99-1]](/bjoc/content/figures/1860-5397-9-99-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Byproducts formed by aminohalogenation of N-tosyloxycarbamate 8 with FeCl2/TMSCl in EtOH (see Table 1; entry 7).
Figure 1: Byproducts formed by aminohalogenation of N-tosyloxycarbamate 8 with FeCl2/TMSCl in EtOH (see Table 1; ent...
The present study on the aminohalogenation reaction of carbamate 8 has inspired mechanistic insights that deserve discussion (Scheme 4). We hypothesize that cyclized material 5a/5b, reduced material 9, and enone 10 are generated from an N–iron complex (i) that has free-radical character, as previously proposed in the catalytic cyclization of azidoformates [30,38-40]. The contrasting yields obtained from N-tosyloxycarbamate 8 and azidoformate 3 under FeBr2/Bu4NBr in EtOH conditions (see Table 1, entry 1 versus Scheme 3) likely originate from the distinct chemical property of the N–iron species (i) generated from each substrate. The possible coordination of tosylate anion to the N–iron after the N–O bond cleavage with FeX2 may have affected the electronic and steric characters of intermediate (i), leading to retardation of the subsequent cyclization. Because of the low cyclization rate, the production of reduced carbamate 9 and enone 10 became pronounced. This is consistent with the observation that the relatively efficient production of cyclized material 5a was observed for azidoformate 3, where N–iron intermediate (i) was free from such interactions. One of the other characteristics found in the present transformations was the incomplete consumption of substrate 8 by lowering FeBr2/Bu4NBr loading (e.g., Table 1, entry 5), which, in turn, enabled the efficient conversion of structurally simple N-tosyloxycarbamates into the corresponding cyclic aminobromides [28]. This poor conversion under conditions of less FeX2/Bu4NX loading may be attributable to the decrease of the concentration of reactive FeX2 through capture with the polar amide functionality of 8.
![[1860-5397-9-99-i4]](/bjoc/content/inline/1860-5397-9-99-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Plausible reaction pathways in the aminohalogenation of N-tosyloxycarbamate 8 with FeX2/Bu4NX.
Scheme 4: Plausible reaction pathways in the aminohalogenation of N-tosyloxycarbamate 8 with FeX2/Bu4NX.
It is speculated that product 9 may be produced by trapping N–iron complex (i) with another FeX2 (i→vii→9), whereas enone 10 is likely to be generated via intramolecular allylic hydrogen abstraction followed by halogen transfer to regenerate iron(II) species (i→iv→v→10) and/or by directly releasing FeX2 (i→iv→vi→10) [41]. However, it is worth discussing the process for yielding 9, which theoretically generates two equivalents of iron(III) species per one equivalent of vii. Given the observation that FeCl3/Bu4NCl gave none of the products shown in Table 1, an iron(III) species possibly generated via the halogen exchange of vii with Bu4NX, if any, no longer has catalytic activity and thus the catalytic cycle is terminated. Therefore, active FeX2 species should somehow be regenerated to maintain the catalysis. One possible pathway that may account for the production of carbamate 9 through the regeneration of FeX2 species is the intermolecular hydrogen abstraction from substrate 8 by N–iron species (i) (Scheme 5). The intermediacy of the intermolecular hydrogen abstraction of N–iron species (i) is supported by the fact that the production of 9 was more pronounced in EtOH having a C–H bond α to the oxygen, which likely served as a hydrogen donor (Table 1, entries 1 and 2). It should be mentioned that reduced material 9 may also be produced by Bu4NBr alone as observed in our previous study [28]. To elucidate the contribution of this pathway, compound 8 was treated with Bu4NX in t-BuOH. However, no reduced material was obtained within the reaction times depicted in Table 1 [for instance, 0.5 h stirring for Bu4NBr (Table 1, entry 3) and 2.5 h stirring for Bu4NCl (Table 1, entry 4)], indicating that the non-iron-mediated process is not significant [42]. Various yields of 9 obtained by loading consistent amounts (1.2–1.5 equiv) of Bu4NX salts also indicated the poor contribution of the pathway. Chan and co-workers demonstrated that an iron–imido complex generated from FeCl2/PhI=NTs underwent radical hydrogen abstraction from a formyl group, and combined the resultant radicals (hydrogen atom abstraction/radical rebound pathway) to provide amides [43,44]. The involvement of such an iron complex (shown in brackets in Scheme 4) that features radical/metal–nitrenoid properties can be considered in our reactions. A recent study by Betley and co-workers on high-spin iron–imido complexes generated by the reactions of alkyl azides with FeCl2 bearing dipyrromethene ligands revealed the radical character of the complex [39,40], harmonizing well with our result, which implies the intermediacy of the nitrogen radical species.
![[1860-5397-9-99-i5]](/bjoc/content/inline/1860-5397-9-99-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible reaction pathway to produce compounds 9 and 10.
Scheme 5: Plausible reaction pathway to produce compounds 9 and 10.
Conclusion
We have developed a new approach to key compounds 5a/5b for (−)-agelastatin A (1) synthesis, which features the iron(II)-mediated radical cyclization of N-tosyloxycarbamate, a safe azidoformate surrogate. Although somewhat moderate chemical yields of the compounds were obtained in this study, the elimination of hazardous synthetic processes enables the establishment of more robust strategies to access 1. Furthermore, the present study has allowed us to obtain mechanistic insights suggesting that N–iron species (i) has a metal-radical character. Much work is currently being undertaken to comprehend fully the unique properties of the present reactions.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data of new compounds, and 1H/13C NMR spectra. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
The authors are deeply indebted to Professor Tetsuaki Tanaka for his encouragement. This work was supported by a Special Grant generously provided by the Hoansha Foundation and a Grant-in-Aid for Scientific Research on Innovative Areas [No. 22136006] from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
References
-
D’Ambrosio, M.; Guerriero, A.; Debitus, C.; Ribes, O.; Pusset, J.; Leroy, S.; Pietra, F. J. Chem. Soc., Chem. Commun. 1993, 1305. doi:10.1039/C39930001305
Return to citation in text: [1] -
Hong, T. W.; Jímenez, D. R.; Molinski, T. F. J. Nat. Prod. 1998, 61, 158. doi:10.1021/np9703813
Return to citation in text: [1] -
Tilvi, S.; Moriou, C.; Martin, M.-T.; Gallard, J.-F.; Sorres, J.; Patel, K.; Petek, S.; Debitus, C.; Ermolenko, L.; Al-Mourabit, A. J. Nat. Prod. 2010, 73, 720. doi:10.1021/np900539j
Return to citation in text: [1] -
Pettit, G. R.; Ducki, S.; Herald, D. L.; Doubek, D. L.; Schmidt, J. M.; Chapuis, J.-C. Oncol. Res. 2005, 15, 11.
Return to citation in text: [1] -
Mason, C. K.; McFarlane, S.; Johnston, P. G.; Crowe, P.; Erwin, P. J.; Domostoj, M. M.; Campbell, F. C.; Manaviazar, S.; Hale, K. J.; El-Tanani, M. Mol. Cancer Ther. 2008, 7, 548. doi:10.1158/1535-7163.MCT-07-2251
Return to citation in text: [1] -
Stien, D.; Anderson, G. T.; Chase, C. E.; Koh, Y.-H.; Weinreb, S. M. J. Am. Chem. Soc. 1999, 121, 9574. doi:10.1021/ja992487l
Return to citation in text: [1] -
Feldman, K. S.; Saunders, J. C. J. Am. Chem. Soc. 2002, 124, 9060. doi:10.1021/ja027121e
Return to citation in text: [1] -
Hale, K. J.; Domostoj, M. M.; Tocher, D. A.; Irving, E.; Scheinmann, F. Org. Lett. 2003, 5, 2927. doi:10.1021/ol035036l
Return to citation in text: [1] -
Domostoj, M. M.; Irving, E.; Scheinmann, F.; Hale, K. J. Org. Lett. 2004, 6, 2615. doi:10.1021/ol0490476
Return to citation in text: [1] -
Davis, F. A.; Deng, J. Org. Lett. 2005, 7, 621. doi:10.1021/ol047634l
Return to citation in text: [1] -
Trost, B. M.; Dong, G. J. Am. Chem. Soc. 2006, 128, 6054. doi:10.1021/ja061105q
Return to citation in text: [1] -
Ichikawa, Y.; Yamaoka, T.; Nakano, K.; Kotsuki, H. Org. Lett. 2007, 9, 2989. doi:10.1021/ol0709735
Return to citation in text: [1] -
Dickson, D. P.; Wardrop, D. J. Org. Lett. 2009, 11, 1341. doi:10.1021/ol900133v
Return to citation in text: [1] -
Wehn, P. M.; Du Bois, J. Angew. Chem., Int. Ed. 2009, 48, 3802. doi:10.1002/anie.200806292
Return to citation in text: [1] -
Hama, N.; Matsuda, T.; Sato, T.; Chida, N. Org. Lett. 2009, 11, 2687. doi:10.1021/ol900799e
Return to citation in text: [1] -
Movassaghi, M.; Siegel, D. S.; Han, S. Chem. Sci. 2010, 1, 561. doi:10.1039/c0sc00351d
Return to citation in text: [1] -
Menjo, Y.; Hamajima, A.; Sasaki, N.; Hamada, Y. Org. Lett. 2011, 13, 5744. doi:10.1021/ol2023054
Return to citation in text: [1] -
Reyes, J. C. P.; Romo, D. Angew. Chem., Int. Ed. 2012, 51, 6870. doi:10.1002/anie.201200959
Return to citation in text: [1] -
Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 7516. doi:10.1021/ja301120z
Return to citation in text: [1] -
Hale, K. J.; Domostoj, M. M.; El-Tanani, M.; Campbell, F. C.; Mason, C. K. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Academic Press: London, 2005; Vol. 6, p 352. doi:10.1016/S1874-6004(05)80034-6
See for selected reviews of agelastatins and related compounds.
Return to citation in text: [1] -
Weinreb, S. M. Nat. Prod. Rep. 2007, 24, 931. doi:10.1039/b700206h
Return to citation in text: [1] -
Dong, G. Pure Appl. Chem. 2010, 82, 2231. doi:10.1351/PAC-CON-10-08-04
Return to citation in text: [1] -
Young, I. S.; Thornton, P. D.; Thompson, A. Nat. Prod. Rep. 2010, 27, 1801. doi:10.1039/c0np00014k
Return to citation in text: [1] -
Al-Mourabit, A.; Zancanella, M. A.; Tilvi, S.; Romo, D. Nat. Prod. Rep. 2011, 28, 1229. doi:10.1039/c0np00013b
Return to citation in text: [1] -
Yamaoka, T.; Ichikawa, Y.; Kotsuki, H. J. Synth. Org. Chem., Jpn. 2012, 70, 615. doi:10.5059/yukigoseikyokaishi.70.615
Return to citation in text: [1] -
Yoshimitsu, T.; Ino, T.; Tanaka, T. Org. Lett. 2008, 10, 5457. doi:10.1021/ol802225g
Return to citation in text: [1] [2] [3] -
Yoshimitsu, T.; Ino, T.; Futamura, N.; Kamon, T.; Tanaka, T. Org. Lett. 2009, 11, 3402. doi:10.1021/ol9012684
Return to citation in text: [1] [2] [3] -
Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363. doi:10.1039/c2ob07190h
Return to citation in text: [1] [2] [3] [4] -
Nguyen, Q.; Nguyen, T.; Driver, T. G. J. Am. Chem. Soc. 2013, 135, 620. doi:10.1021/ja3113565
See for a recent application of catalytic FeBr2 to C–H amination.
Return to citation in text: [1] -
Bach, T.; Schlummer, B.; Harms, K. Chem. Commun. 2000, 287. doi:10.1039/a909009f
Return to citation in text: [1] [2] [3] -
Bach, T.; Schlummer, B.; Harms, K. Synlett 2000, 1330. doi:10.1055/s-2000-7129
Return to citation in text: [1] [2] -
Bach, T.; Schlummer, B.; Harms, K. Chem.–Eur. J. 2001, 7, 2581. doi:10.1002/1521-3765(20010618)7:12<2581::AID-CHEM25810>3.0.CO;2-O
Return to citation in text: [1] [2] -
Churchill, D. G.; Rojas, C. M. Tetrahedron Lett. 2002, 43, 7225. doi:10.1016/S0040-4039(02)01658-1
Return to citation in text: [1] [2] -
Bacci, J. P.; Greenman, K. L.; Van Vranken, D. L. J. Org. Chem. 2003, 68, 4955. doi:10.1021/jo0340410
Return to citation in text: [1] [2] -
Danielec, H.; Klügge, J.; Schlummer, B.; Bach, T. Synthesis 2004, 551. doi:10.1055/s-2005-918500
Return to citation in text: [1] [2] -
Lebel, H.; Huard, K.; Lectard, S. J. Am. Chem. Soc. 2005, 127, 14198. doi:10.1021/ja0552850
Return to citation in text: [1] -
When enone 10 was treated with TMSCl in EtOH at room temperature, diethylketal 12 (19%) and ketone 11 (10%) were generated. The TLC analysis of the reaction mixture clearly showed that diethylketal 12 was an initial product, which gradually underwent decomposition to give ketone 11.
Return to citation in text: [1] -
Protonated cation radical x is considered to be produced immediately after N–O cleavage and may be responsible for the distinct reactivities. However, because of its high acidity, the proton on the carbamoyl nitrogen atom of x likely dissociates to provide intermediate i.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-99-i7.svg?max-width=637&scale=1.18182)
Return to citation in text: [1] -
King, E. R.; Hennessy, E. T.; Betley, T. A. J. Am. Chem. Soc. 2011, 133, 4917. doi:10.1021/ja110066j
See for recent studies on C–H cleavage by N–iron complex showing radical character.
Return to citation in text: [1] [2] -
Paradine, S. M.; White, M. C. J. Am. Chem. Soc. 2012, 134, 2036. doi:10.1021/ja211600g
and references cited therein.
Return to citation in text: [1] [2] -
It can also be assumed that enone 10 is generated from intermediate iv via direct fragmentation of an isocyanate and iron(II) halide. We thank one of the referees for suggesting such a possibility.
Return to citation in text: [1] -
Treatment of substrate 8 with Bu4NBr for 2.5 h gave a small amount of 9 (9%) along with unreacted 8 (74%), whose chemical yields were estimated by 1H NMR analysis of the crude mixture. In contrast, no reduced product 9 was generated at all with Bu4NCl after 2.5 h. In this case, after 5.5 h, carbamate 9 was formed in 3% yield accompanying the recovery of 8 (85%).
Return to citation in text: [1] -
Ton, T. M. U.; Tejo, C.; Tania, S.; Chang, J. W. W.; Chan, P. W. H. J. Org. Chem. 2011, 76, 4894. doi:10.1021/jo200284a
Return to citation in text: [1] -
Maestre, L.; Sameera, W. M. C.; Mar Díaz-Requejo, M.; Maseras, F.; Pérez, P. J. J. Am. Chem. Soc. 2013, 135, 1338. doi:10.1021/ja307229e
See for a recent discussion on the mechanisms of metal–nitrenide catalysis.
Return to citation in text: [1]
| 43. | Ton, T. M. U.; Tejo, C.; Tania, S.; Chang, J. W. W.; Chan, P. W. H. J. Org. Chem. 2011, 76, 4894. doi:10.1021/jo200284a |
| 44. |
Maestre, L.; Sameera, W. M. C.; Mar Díaz-Requejo, M.; Maseras, F.; Pérez, P. J. J. Am. Chem. Soc. 2013, 135, 1338. doi:10.1021/ja307229e
See for a recent discussion on the mechanisms of metal–nitrenide catalysis. |
| 28. | Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363. doi:10.1039/c2ob07190h |
| 42. | Treatment of substrate 8 with Bu4NBr for 2.5 h gave a small amount of 9 (9%) along with unreacted 8 (74%), whose chemical yields were estimated by 1H NMR analysis of the crude mixture. In contrast, no reduced product 9 was generated at all with Bu4NCl after 2.5 h. In this case, after 5.5 h, carbamate 9 was formed in 3% yield accompanying the recovery of 8 (85%). |
| 1. | D’Ambrosio, M.; Guerriero, A.; Debitus, C.; Ribes, O.; Pusset, J.; Leroy, S.; Pietra, F. J. Chem. Soc., Chem. Commun. 1993, 1305. doi:10.1039/C39930001305 |
| 2. | Hong, T. W.; Jímenez, D. R.; Molinski, T. F. J. Nat. Prod. 1998, 61, 158. doi:10.1021/np9703813 |
| 3. | Tilvi, S.; Moriou, C.; Martin, M.-T.; Gallard, J.-F.; Sorres, J.; Patel, K.; Petek, S.; Debitus, C.; Ermolenko, L.; Al-Mourabit, A. J. Nat. Prod. 2010, 73, 720. doi:10.1021/np900539j |
| 4. | Pettit, G. R.; Ducki, S.; Herald, D. L.; Doubek, D. L.; Schmidt, J. M.; Chapuis, J.-C. Oncol. Res. 2005, 15, 11. |
| 5. | Mason, C. K.; McFarlane, S.; Johnston, P. G.; Crowe, P.; Erwin, P. J.; Domostoj, M. M.; Campbell, F. C.; Manaviazar, S.; Hale, K. J.; El-Tanani, M. Mol. Cancer Ther. 2008, 7, 548. doi:10.1158/1535-7163.MCT-07-2251 |
| 26. | Yoshimitsu, T.; Ino, T.; Tanaka, T. Org. Lett. 2008, 10, 5457. doi:10.1021/ol802225g |
| 28. | Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363. doi:10.1039/c2ob07190h |
| 27. | Yoshimitsu, T.; Ino, T.; Futamura, N.; Kamon, T.; Tanaka, T. Org. Lett. 2009, 11, 3402. doi:10.1021/ol9012684 |
| 41. | It can also be assumed that enone 10 is generated from intermediate iv via direct fragmentation of an isocyanate and iron(II) halide. We thank one of the referees for suggesting such a possibility. |
| 26. | Yoshimitsu, T.; Ino, T.; Tanaka, T. Org. Lett. 2008, 10, 5457. doi:10.1021/ol802225g |
| 37. | When enone 10 was treated with TMSCl in EtOH at room temperature, diethylketal 12 (19%) and ketone 11 (10%) were generated. The TLC analysis of the reaction mixture clearly showed that diethylketal 12 was an initial product, which gradually underwent decomposition to give ketone 11. |
| 6. | Stien, D.; Anderson, G. T.; Chase, C. E.; Koh, Y.-H.; Weinreb, S. M. J. Am. Chem. Soc. 1999, 121, 9574. doi:10.1021/ja992487l |
| 7. | Feldman, K. S.; Saunders, J. C. J. Am. Chem. Soc. 2002, 124, 9060. doi:10.1021/ja027121e |
| 8. | Hale, K. J.; Domostoj, M. M.; Tocher, D. A.; Irving, E.; Scheinmann, F. Org. Lett. 2003, 5, 2927. doi:10.1021/ol035036l |
| 9. | Domostoj, M. M.; Irving, E.; Scheinmann, F.; Hale, K. J. Org. Lett. 2004, 6, 2615. doi:10.1021/ol0490476 |
| 10. | Davis, F. A.; Deng, J. Org. Lett. 2005, 7, 621. doi:10.1021/ol047634l |
| 11. | Trost, B. M.; Dong, G. J. Am. Chem. Soc. 2006, 128, 6054. doi:10.1021/ja061105q |
| 12. | Ichikawa, Y.; Yamaoka, T.; Nakano, K.; Kotsuki, H. Org. Lett. 2007, 9, 2989. doi:10.1021/ol0709735 |
| 13. | Dickson, D. P.; Wardrop, D. J. Org. Lett. 2009, 11, 1341. doi:10.1021/ol900133v |
| 14. | Wehn, P. M.; Du Bois, J. Angew. Chem., Int. Ed. 2009, 48, 3802. doi:10.1002/anie.200806292 |
| 15. | Hama, N.; Matsuda, T.; Sato, T.; Chida, N. Org. Lett. 2009, 11, 2687. doi:10.1021/ol900799e |
| 16. | Movassaghi, M.; Siegel, D. S.; Han, S. Chem. Sci. 2010, 1, 561. doi:10.1039/c0sc00351d |
| 17. | Menjo, Y.; Hamajima, A.; Sasaki, N.; Hamada, Y. Org. Lett. 2011, 13, 5744. doi:10.1021/ol2023054 |
| 18. | Reyes, J. C. P.; Romo, D. Angew. Chem., Int. Ed. 2012, 51, 6870. doi:10.1002/anie.201200959 |
| 19. | Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 7516. doi:10.1021/ja301120z |
| 20. |
Hale, K. J.; Domostoj, M. M.; El-Tanani, M.; Campbell, F. C.; Mason, C. K. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Academic Press: London, 2005; Vol. 6, p 352. doi:10.1016/S1874-6004(05)80034-6
See for selected reviews of agelastatins and related compounds. |
| 21. | Weinreb, S. M. Nat. Prod. Rep. 2007, 24, 931. doi:10.1039/b700206h |
| 22. | Dong, G. Pure Appl. Chem. 2010, 82, 2231. doi:10.1351/PAC-CON-10-08-04 |
| 23. | Young, I. S.; Thornton, P. D.; Thompson, A. Nat. Prod. Rep. 2010, 27, 1801. doi:10.1039/c0np00014k |
| 24. | Al-Mourabit, A.; Zancanella, M. A.; Tilvi, S.; Romo, D. Nat. Prod. Rep. 2011, 28, 1229. doi:10.1039/c0np00013b |
| 25. | Yamaoka, T.; Ichikawa, Y.; Kotsuki, H. J. Synth. Org. Chem., Jpn. 2012, 70, 615. doi:10.5059/yukigoseikyokaishi.70.615 |
| 30. | Bach, T.; Schlummer, B.; Harms, K. Chem. Commun. 2000, 287. doi:10.1039/a909009f |
| 38. |
Protonated cation radical x is considered to be produced immediately after N–O cleavage and may be responsible for the distinct reactivities. However, because of its high acidity, the proton on the carbamoyl nitrogen atom of x likely dissociates to provide intermediate i.
|
| 39. |
King, E. R.; Hennessy, E. T.; Betley, T. A. J. Am. Chem. Soc. 2011, 133, 4917. doi:10.1021/ja110066j
See for recent studies on C–H cleavage by N–iron complex showing radical character. |
| 40. |
Paradine, S. M.; White, M. C. J. Am. Chem. Soc. 2012, 134, 2036. doi:10.1021/ja211600g
and references cited therein. |
| 26. | Yoshimitsu, T.; Ino, T.; Tanaka, T. Org. Lett. 2008, 10, 5457. doi:10.1021/ol802225g |
| 27. | Yoshimitsu, T.; Ino, T.; Futamura, N.; Kamon, T.; Tanaka, T. Org. Lett. 2009, 11, 3402. doi:10.1021/ol9012684 |
| 28. | Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363. doi:10.1039/c2ob07190h |
| 30. | Bach, T.; Schlummer, B.; Harms, K. Chem. Commun. 2000, 287. doi:10.1039/a909009f |
| 31. | Bach, T.; Schlummer, B.; Harms, K. Synlett 2000, 1330. doi:10.1055/s-2000-7129 |
| 32. | Bach, T.; Schlummer, B.; Harms, K. Chem.–Eur. J. 2001, 7, 2581. doi:10.1002/1521-3765(20010618)7:12<2581::AID-CHEM25810>3.0.CO;2-O |
| 33. | Churchill, D. G.; Rojas, C. M. Tetrahedron Lett. 2002, 43, 7225. doi:10.1016/S0040-4039(02)01658-1 |
| 34. | Bacci, J. P.; Greenman, K. L.; Van Vranken, D. L. J. Org. Chem. 2003, 68, 4955. doi:10.1021/jo0340410 |
| 35. | Danielec, H.; Klügge, J.; Schlummer, B.; Bach, T. Synthesis 2004, 551. doi:10.1055/s-2005-918500 |
| 30. | Bach, T.; Schlummer, B.; Harms, K. Chem. Commun. 2000, 287. doi:10.1039/a909009f |
| 31. | Bach, T.; Schlummer, B.; Harms, K. Synlett 2000, 1330. doi:10.1055/s-2000-7129 |
| 32. | Bach, T.; Schlummer, B.; Harms, K. Chem.–Eur. J. 2001, 7, 2581. doi:10.1002/1521-3765(20010618)7:12<2581::AID-CHEM25810>3.0.CO;2-O |
| 33. | Churchill, D. G.; Rojas, C. M. Tetrahedron Lett. 2002, 43, 7225. doi:10.1016/S0040-4039(02)01658-1 |
| 34. | Bacci, J. P.; Greenman, K. L.; Van Vranken, D. L. J. Org. Chem. 2003, 68, 4955. doi:10.1021/jo0340410 |
| 35. | Danielec, H.; Klügge, J.; Schlummer, B.; Bach, T. Synthesis 2004, 551. doi:10.1055/s-2005-918500 |
| 28. | Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363. doi:10.1039/c2ob07190h |
| 29. |
Nguyen, Q.; Nguyen, T.; Driver, T. G. J. Am. Chem. Soc. 2013, 135, 620. doi:10.1021/ja3113565
See for a recent application of catalytic FeBr2 to C–H amination. |
| 39. |
King, E. R.; Hennessy, E. T.; Betley, T. A. J. Am. Chem. Soc. 2011, 133, 4917. doi:10.1021/ja110066j
See for recent studies on C–H cleavage by N–iron complex showing radical character. |
| 40. |
Paradine, S. M.; White, M. C. J. Am. Chem. Soc. 2012, 134, 2036. doi:10.1021/ja211600g
and references cited therein. |
| 27. | Yoshimitsu, T.; Ino, T.; Futamura, N.; Kamon, T.; Tanaka, T. Org. Lett. 2009, 11, 3402. doi:10.1021/ol9012684 |
| 36. | Lebel, H.; Huard, K.; Lectard, S. J. Am. Chem. Soc. 2005, 127, 14198. doi:10.1021/ja0552850 |
© 2013 Shigeoka et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)