Search results
Search for "molecular dynamics" in Full Text gives 109 result(s) in Beilstein Journal of Organic Chemistry.
6’-Fluoro[4.3.0]bicyclo nucleic acid: synthesis, biophysical properties and molecular dynamics simulations
Beilstein J. Org. Chem. 2018, 14, 3088–3097, doi:10.3762/bjoc.14.288

- = −1.5 to −3.7 °C). Molecular dynamics simulation on the nucleoside and oligonucleotide level revealed the preference of the C1’-exo/C2’-endo alignment of the furanose ring. Moreover, the simulation of duplexes with complementary RNA disclosed a DNA/RNA-type duplex structure suggesting that this
- modification might be a substrate for RNase H. Keywords: DNA/RNA affinity; fluorinated cyclopropanes; fluorinated nucleic acids; molecular dynamics simulations; sugar modified nucleosides; Introduction A powerful strategy for the treatment of various disorders like cancer, viral and inherited diseases is the
- unit and possibly positively impact the duplex stability. Here we report on the synthesis of the two 6’F-bc4,3 pyrimidine analogs with the base T and C, their incorporation into DNA, their biophysical properties, as well as a structural analysis by molecular dynamics simulations of hybrid DNA and RNA









Diazirine-functionalized mannosides for photoaffinity labeling: trouble with FimH
Beilstein J. Org. Chem. 2018, 14, 1890–1900, doi:10.3762/bjoc.14.163

- X-ray crystallography, studies in solution add valuable information in molecular recognition studies as they take molecular dynamics as well as solvent effects into consideration. In the latter respect, photoaffinity labeling has evolved as a useful tool for studies under physiological conditions [1








Synthesis and photophysical studies of a multivalent photoreactive RuII-calix[4]arene complex bearing RGD-containing cyclopentapeptides
Beilstein J. Org. Chem. 2018, 14, 1758–1768, doi:10.3762/bjoc.14.150

- presented in Figure 2, as issued from a molecular dynamics (MD) simulations. The ruthenium complex and the RGD units are spatially well-separated thanks to their grafting on opposite faces of the rigid calixarene-based platform. In this conformation, the distances between the Ru atom and each of the nearest









An overview of recent advances in duplex DNA recognition by small molecules
Beilstein J. Org. Chem. 2018, 14, 1051–1086, doi:10.3762/bjoc.14.93
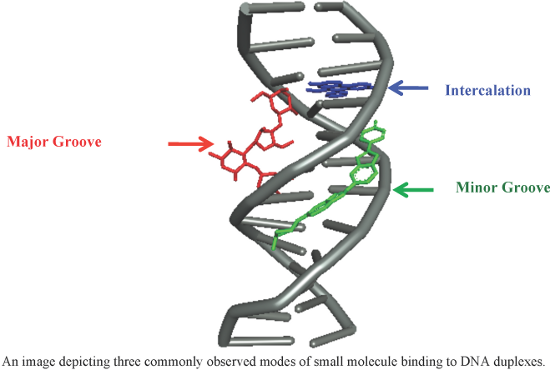
- binding affinity using sophisticated molecular dynamics approach with eight different nucleotides having variety of sequences. It has been observed that QUE appears to be a minor groove binder, whereas FLP involves in combined mode of interaction such as minor groove binding and intercalation. A set of






















Crystal structure of the inclusion complex of cholesterol in β-cyclodextrin and molecular dynamics studies
Beilstein J. Org. Chem. 2018, 14, 838–848, doi:10.3762/bjoc.14.69

- inclusion compound remains very stable in aqueous solution at both temperatures. Keywords: beta-cyclodextrin; cholesterol; crystal structure; molecular dynamics; Introduction Cholesterol ((3β)-cholest-5-en-3-ol, CHL, Figure 1a) is a polycyclic steroid that is synthesized in mammalian cells and has a
- supported by the above mentioned guest–host interactions. Therefore, cholesterol is always found in the cavity of the β-CD dimer, its size and shape prohibiting its diffusion in the crystal. Molecular dynamics The crystallographically determined atomic coordinates of the CHL/β-CD complex (host/guest
- stoichiometry 2:1) (Figure 2a) were subjected to equilibration and subsequent molecular dynamics simulations at both 300 and 340 K in explicit water solvent for almost 12 ns with the aim to monitor the dynamic behavior of CHL in β-CD in two different temperatures, study the host–guest interactions during the































Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids – tutorial
Beilstein J. Org. Chem. 2018, 14, 84–105, doi:10.3762/bjoc.14.5
















The use of 4,4,4-trifluorothreonine to stabilize extended peptide structures and mimic β-strands
Beilstein J. Org. Chem. 2017, 13, 2842–2853, doi:10.3762/bjoc.13.276

- according to their central fluorinated or non-fluorinated residue, all-atom molecular dynamics (MD) simulations were performed using the GROMACS 4.5 package, with the OPLS-AA force field in combination with the SPC/E water model (for a complete description of the method, see Supporting Information File 1
- obtained through the aldol reaction of trifluoroacetaldehyde with the Ni(II) complex of the chiral Schiff base of glycine. The conformational analysis of these pentapeptides was conducted by the combined use of NMR spectroscopy and molecular dynamics simulations. NMR conformational studies showed that the
- , L-threonine, (2S,3R)-L-CF3-threonine and (2S,3S)-L-CF3-threonine were prepared. The capacity of (2S,3S)- and (2S,3R)-CF3-threonine analogues to stabilize an extended structure when introduced in the central position of pentapeptides is demonstrated by NMR conformational studies and molecular








What contributes to an effective mannose recognition domain?
Beilstein J. Org. Chem. 2017, 13, 2584–2595, doi:10.3762/bjoc.13.255

- well as the influence of highly mobile vs conserved waters were analyzed. For the assessment of the dynamic behavior of the ligand complexes of the seven calcium-dependent lectins, 20 ns molecular dynamics (MD) simulations were performed [57]. The most prominent interactions of O–C3 and O–C4 of the









2-Methyl-2,4-pentanediol (MPD) boosts as detergent-substitute the performance of ß-barrel hybrid catalyst for phenylacetylene polymerization
Beilstein J. Org. Chem. 2017, 13, 1498–1506, doi:10.3762/bjoc.13.148

- rhodium-based biohybrid catalyst. Unlike commonly used detergents such as sodium dodecyl sulfate or polyethylene polyethyleneglycol, MPD does not form micelles in solution. Molecular dynamics simulations revealed the effect and position of stabilizing MPD molecules. The advantage of the amphiphilic MPD
- carried out, reaching higher molecular weights and yields compared to catalysis with the micelle-forming refolding reagent PE–PEG. Minimum of MPD molecules was analyzed by molecular dynamics studies to enable refolding of SDS-denatured transmembrane protein FhuA ΔCVFtev [29]. This report aims to
- 1). Molecular dynamics (MD) simulations reveal an optimal minimum number of ≈200 MPD molecules for shielding the hydrophobic transmembrane region of FhuA ΔCVFtev MD simulations of FhuA ΔCVFtev were performed in a box with varying numbers of MPD molecules from 126 MPD, 189 MPD, 252 MPD to 378 MPD






Aqueous semisynthesis of C-glycoside glycamines from agarose
Beilstein J. Org. Chem. 2017, 13, 1222–1229, doi:10.3762/bjoc.13.121

- aminomonosaccharides 9 and 13 are interesting moieties, regarding their resemblance with the bioactive (+)-muscarine 14 (Figure 2). Indeed, ongoing docking and molecular dynamics experiments revealed the amino-AnGal moiety as a promising platform to launch the design of new mAChR modulators [11]. The differences in





G-Protein coupled receptors: answers from simulations
Beilstein J. Org. Chem. 2017, 13, 1071–1078, doi:10.3762/bjoc.13.106

- Timothy Clark Computer-Chemie-Centrum, Department of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nuernberg, Naegelsbachstr. 25, 91052 Erlangen, Germany 10.3762/bjoc.13.106 Abstract Molecular-dynamics (MD) simulations are playing an increasingly important role in research into
- ; molecular dynamics; Introduction Evolution is a unique optimization mechanism. Firstly, it stops optimizing as soon as an acceptable solution is reached. There is no evolutionary pressure for elegance, simplicity or even effectiveness above the critical threshold. Secondly, because evolution always starts
- experimental findings [29] suggest that both an agonist ligand and a bound G-protein are necessary in order to activate GPCRs. It is therefore significant that the first molecular dynamics (MD) simulations of a ternary GPCR complex were reported only four years ago [30]. Such simulations are now commonplace





Continuous-flow processes for the catalytic partial hydrogenation reaction of alkynes
Beilstein J. Org. Chem. 2017, 13, 734–754, doi:10.3762/bjoc.13.73

- selectivity trend was explained in terms of both adsorption mode on and relative accessibility to Pd active sites, depending on surface potentials and hindrance of modifiers, on the basis of density functional theory and molecular dynamics calculations. The rationale was summarized in the so-called


















Membrane properties of hydroxycholesterols related to the brain cholesterol metabolism
Beilstein J. Org. Chem. 2017, 13, 720–727, doi:10.3762/bjoc.13.71

- fluorescence techniques [21]. Also, a decreased but still significant effect of the hydroxysterols on lipid condensation compared to native cholesterol was found in molecular dynamics simulations, which is probably caused by an increased tilt angle of the sterols to the membrane normal [8][21]. However, using





Inclusion complexes of β-cyclodextrin with tricyclic drugs: an X-ray diffraction, NMR and molecular dynamics study
Beilstein J. Org. Chem. 2017, 13, 714–719, doi:10.3762/bjoc.13.70

- /β-CD and 2/β-CD complexes, with the aromatic ring system entering the cavity from the large rim of the cyclodextrin and the alkylammonium chain protruding out of the cavity and facing the secondary OH rim. These features matched those found in the molecular dynamics (MD) simulations in solution and
- . Keywords: amitriptyline; β-cyclodextrin; crystal structure; cyclobenzaprine; molecular dynamics simulations; NOE; Introduction The present paper reports on a multidisciplinary approach [1][2] based on single crystal X-ray diffraction, solution NMR spectroscopy and molecular dynamics (MD) simulations with
- : 2K points acquired in the F2 domain, 512 increments and subsequent zero-filling to 1K to process data. Molecular dynamics simulations The simulations employed InsightII/Discover [15] with the CVFF force field [16]. The structure of molecules 1 and 2 were first subjected to an MD run in vacuo and








Computational methods in drug discovery
Beilstein J. Org. Chem. 2016, 12, 2694–2718, doi:10.3762/bjoc.12.267

- , agonists, inhibitors, etc. of a target) design. Molecular dynamics (MD) simulations are frequently used in SBDD to give insights into not only how ligands bind with target proteins but also the pathways of interaction and to account for target flexibility. This is especially important when drug targets are
- AMBER [120] (Assisted Model Building and Energy Refinement) which have been built mainly for molecular dynamics simulations. The molecular docking program DOCK [121] uses force-field based scoring functions derived from molecular dynamics force-field AMBER. Empirical scoring functions Empirical scoring
- , these fragments can be joined to obtain a possible new drug molecule. In the SILCS (site identification by ligand competitive saturation) method, molecular dynamics simulations are used to identify fragments that bind to a target [161][162]. SILCS uses explicit molecular dynamics simulations where the










Dynamic behavior of rearranging carbocations – implications for terpene biosynthesis
Beilstein J. Org. Chem. 2016, 12, 377–390, doi:10.3762/bjoc.12.41

- [31][32][33][34]. To acquire evidence for non-statistical dynamic effects, molecular dynamics (MD) simulations are run for a statistically relevant number of trajectories (typically on the order of hundreds or thousands, depending on the system and the starting point for trajectories) [35][36]. The
- –Oppenheimer Molecular Dynamics (BOMD) calculations, so that nuclear motion and electronic structure are calculated separately, the former propagated classically and the latter determined using quantum mechanics. As with any computational (or experimental) study, there will always be a tradeoff between
- commonly used [35][36][37][38][39][40]. In particular, the B3LYP and mPW1PW91 functionals, along with small to medium sized basis sets have seen the most use in studying carbocation rearrangements of relevance to biosynthesis [6]. Using molecular dynamics trajectories to rationalize experimental results is












Aggregation behavior of amphiphilic cyclodextrins in a nonpolar solvent: evidence of large-scale structures by atomistic molecular dynamics simulations and solution studies
Beilstein J. Org. Chem. 2016, 12, 73–80, doi:10.3762/bjoc.12.8

- have been usually investigated and characterized in water for their potential use as nanocarriers for drug delivery, but they can also aggregate in apolar solvents, as shown in the present paper through atomistic molecular dynamics simulations and dynamic light scattering measurements. The simulations
- nanoaggregates even in apolar solvents. Keywords: aggregation; amphiphilic cyclodextrins; molecular dynamics; nanoparticles; self-assembly; simulations; Introduction Amphiphilic cyclodextrins (aCD) are a class of molecules highly investigated for their self-assembly properties and inherent potential
- shown in Scheme 1b (n = 1), dissolved in an apolar solvent and an atomistic Molecular Dynamics study of a model compound (n = 0 in Scheme 1) carried out in vacuo. In particular, the latter approach aims to model the bottom-up aggregation of the model aCD molecules, an approach that was started in [11









Polydisperse methyl β-cyclodextrin–epichlorohydrin polymers: variable contact time 13C CP-MAS solid-state NMR characterization
Beilstein J. Org. Chem. 2015, 11, 2785–2794, doi:10.3762/bjoc.11.299

- information (the chemical connectivity) and dynamic insights (the overall molecular dynamics in the solid state). Pines et al. [38] were the first to discuss the effect of different types of motion – such as molecular conformational changes, molecular reorientation and macroscopic sample rotation – on the CP










Physical properties and biological activities of hesperetin and naringenin in complex with methylated β-cyclodextrin
Beilstein J. Org. Chem. 2015, 11, 2763–2773, doi:10.3762/bjoc.11.297

- hesperetin with cyclodextrins (β-CD and DM-β-CD) were theoretically investigated by molecular dynamics simulation. The free energy values obtained suggested a more stable inclusion complex with DM-β-CD. The vdW force is the main guest–host interaction when hesperetin binds with CDs. The phase solubility
- physical properties and biological activities of hesperetin and naringenin through complexation with cyclodextrins. Computational tools (molecular dynamics simulation) were adopted to first predict the stability of flavanones/CDs inclusion complexes. Consequently, the experimental phase solubility and
- values showed the same trend with values from molecular dynamics simulation that complexing with DM-β-CD was more effective than with β-CD, and the values obtained were in good agreement with the previous report [42]. These results suggested that both flavanones bind to and interact with DM-β-CD stronger








Aggregation behaviour of amphiphilic cyclodextrins: the nucleation stage by atomistic molecular dynamics simulations
Beilstein J. Org. Chem. 2015, 11, 2459–2473, doi:10.3762/bjoc.11.267

- report a theoretical study of the aggregation of a few amphiphilic cyclodextrins carrying hydrophobic thioalkyl groups and hydrophilic ethylene glycol moieties at opposite rims, focusing on the initial nucleation stage in an apolar solvent and in water. The study is based on atomistic molecular dynamics
- correlating their structures with the pharmaceutical properties. Keywords: aggregation; amphiphilic cyclodextrins; micelles; molecular dynamics simulations; nanoparticles; self-assembly; Introduction Inclusion complexes with supramolecular structures formed by native or modified cyclodextrins (CDs) are
- supramolecular aggregates was seldom, if ever, considered, apart from the above-mentioned reference [35]. To improve our understanding of the factors driving the formation of aCD molecular assemblies, we describe in this paper an atomistic molecular dynamics investigation of a model compound of a non-ionic aCD















Co-solvation effect on the binding mode of the α-mangostin/β-cyclodextrin inclusion complex
Beilstein J. Org. Chem. 2015, 11, 2306–2317, doi:10.3762/bjoc.11.251

- anion affinity and selectivity of a neutral anion receptor, bis(cyclopeptide) [17]. Molecular dynamics (MD) simulations can give important insights into the energetics of structural interactions. The hydrated structure of β-CD in aqueous solution [18] and those showing host–guest interactions between
- given in Table 1. Details of molecular dynamics simulations In the present study, all MD simulations were performed using the SANDER module of the Amber10 software package in accordance with the recently reported MD simulations of flavonoid/β-CD inclusion complexes in water [34][35]. The particle-mesh








A comprehensive study of olefin metathesis catalyzed by Ru-based catalysts
Beilstein J. Org. Chem. 2015, 11, 1767–1780, doi:10.3762/bjoc.11.192

- bond of C2H4 is nearly perpendicular to the Ru–methylidene bond, whereas in the bigger substrate the tether forces the coordinated C=C bond to be almost aligned with the Ru–alkylidene bond. However, the molecular dynamics simulations (vide infra) clearly indicate that in the trans geometries the C2H4













Peptide–polymer ligands for a tandem WW-domain, an adaptive multivalent protein–protein interaction: lessons on the thermodynamic fitness of flexible ligands
Beilstein J. Org. Chem. 2015, 11, 837–847, doi:10.3762/bjoc.11.93

- precipitation. Experimental results were compared with parameters obtained from molecular dynamics simulations in order to understand the observed differences between the three carrier materials. In summary, the more rigid and condensed peptide–polymer conjugates based on the dextran scaffold seem to be
- understanding of structure–activity relationships of polymeric ligands. For this purpose, the thermodynamics and the stoichiometry of protein binding events were determined experimentally for all multivalent ligands. Finally, atomistic molecular dynamics simulations were conducted in order to rationalize the
- bivalent binding mode for the complex of Dex-2 and tandem-WW-FBP21, which is supported also by the solubility of the non-crosslinked peptide-polymer–protein complex. Molecular dynamics simulations of multivalent ligands In order to better understand our experimental observations regarding binding







Synthesis of carbohydrate-scaffolded thymine glycoconjugates to organize multivalency
Beilstein J. Org. Chem. 2015, 11, 668–674, doi:10.3762/bjoc.11.75

- [6]. This work is based on the idea that changes of ligand orientation as well as changes of their conformational availability are regulating parameters in carbohydrate recognition, in particular on the cell surface. Indeed, we have formerly shown that the molecular dynamics of glycodendrimers





