Search results
Search for "simulations" in Full Text gives 139 result(s) in Beilstein Journal of Organic Chemistry.
Tetrathiafulvalene – a redox-switchable building block to control motion in mechanically interlocked molecules
Beilstein J. Org. Chem. 2018, 14, 2163–2185, doi:10.3762/bjoc.14.190

- hydrogen bonding. Detailed electrochemical measurements and digital simulations revealed the ring still to be bound to the ammonium station in the TTF●+ state. However, after double oxidation a wheel distribution of 1:1 between the ammonium and the isoxazole station was found indicating a dynamic motion




























Coordination-driven self-assembly vs dynamic covalent chemistry: versatile methods for the synthesis of molecular metallarectangles
Beilstein J. Org. Chem. 2018, 14, 2027–2034, doi:10.3762/bjoc.14.178

- ). Notably, the isolated yields of the metallarectangles are higher than the overall yields of the two-step method. Since attempts to obtain X-ray quality single crystals of the target metallarectangles were unsuccessful, molecular simulations were performed to gain further insight into the structures of the






Synthesis and photophysical studies of a multivalent photoreactive RuII-calix[4]arene complex bearing RGD-containing cyclopentapeptides
Beilstein J. Org. Chem. 2018, 14, 1758–1768, doi:10.3762/bjoc.14.150

- attributed to [9 + H]3+, [9 + 2H]4+ and [9 + 3H]5+ by comparison between the experimental and theoretical isotope distributions (see Supporting Information File 1). Molecular modeling simulations were carried out to provide insights into the size and morphology of conjugate 9. An optimized geometry is
- presented in Figure 2, as issued from a molecular dynamics (MD) simulations. The ruthenium complex and the RGD units are spatially well-separated thanks to their grafting on opposite faces of the rigid calixarene-based platform. In this conformation, the distances between the Ru atom and each of the nearest
- carbon atoms of RGDfK units exceed 30 Å. Along the MD simulations, we noticed that the Ru complex remained far from the cyclic pentapeptides. This is due to the fact that the linkers of each arm are smaller than the size of the calixarene platform, preventing contacts between the Ru complex and the RGDfK









Host–guest complexes of conformationally flexible C-hexyl-2-bromoresorcinarene and aromatic N-oxides: solid-state, solution and computational studies
Beilstein J. Org. Chem. 2018, 14, 1723–1733, doi:10.3762/bjoc.14.146

- conformer space in these simulations, no constraints were enforced on either N-oxide or acetone molecules. The low energy structures obtained from these OPLS-2005 searches were then further analysed using DFT-based techniques [45][46][47]. The resulting optimised geometries of the 3@BrC2, 3@BrC3 and 3@BrC6






The phenyl vinyl ether–methanol complex: a model system for quantum chemistry benchmarking
Beilstein J. Org. Chem. 2018, 14, 1642–1654, doi:10.3762/bjoc.14.140

- in black is compared with simulations, based on fitted parameters that can be assigned to the OH–O’ isomer (complex 1, red) for the PVE–MeOHcomplex. The observed complex has a clear splitting pattern due to the internal rotation of the methyl group of methanol, labeled with A and E. The experimental







Are dispersion corrections accurate outside equilibrium? A case study on benzene
Beilstein J. Org. Chem. 2018, 14, 1181–1191, doi:10.3762/bjoc.14.99

- interest in how to accurately model them. Multiple families of approaches for including dispersion forces in quantum chemical simulations now exist, mostly based around the principle of improving density functional theory (DFT) calculations (see, e.g., some key and recent summaries [3][5][13][14]) through





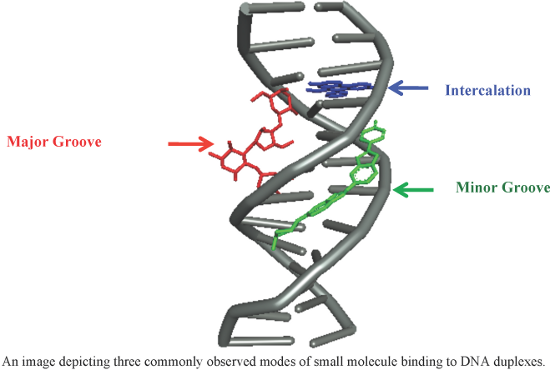
An overview of recent advances in duplex DNA recognition by small molecules
Beilstein J. Org. Chem. 2018, 14, 1051–1086, doi:10.3762/bjoc.14.93






















Correlation effects and many-body interactions in water clusters
Beilstein J. Org. Chem. 2018, 14, 979–991, doi:10.3762/bjoc.14.83

- sound models of the interaction potential for water to be used in molecular simulations. In particular, the role of many-body interactions beyond the two-body interactions, which are often not explicitly taken into account by empirical force fields, can be accurately described by quantum chemistry
- explicitly or implicitly influenced by a water environment. An example for this is the hydrogen-bond cooperativity effect that can have a significant impact on the properties of the bare solute molecules [1]. In order to describe such phenomena, computer simulations have become an indispensable tool, since
- they enable a description of water on a molecular level that often can provide further insights than are accessible from spectroscopic measurements. The basis for such simulations are the so-called force fields that describe both the covalent as well as the noncovalent interactions within the system












Crystal structure of the inclusion complex of cholesterol in β-cyclodextrin and molecular dynamics studies
Beilstein J. Org. Chem. 2018, 14, 838–848, doi:10.3762/bjoc.14.69

- crystalline state comprise the asymmetric unit of the structure. The dimers are arranged in an intermediate (IM) channel packing mode in the crystal. Moreover, MD simulations, at 300 and 340 K, based on the crystallographically determined coordinates of the complex show that the formed cholesterol/β-CD
- characterization of the cholesterol/β-CD inclusion complex [22], its binding affinity [23][24][25], the inclusion mode of the complex [26] and its dynamic behavior through MD simulations [27][28][29] but its crystal structure is absent. In this work, the structure of CHL/β-CD is determined by X-ray crystallography
- and its geometrical features are examined thoroughly. In order to examine the stability of the crystallographically determined model excluding the crystal contacts observed in the crystalline state, MD simulations of the inclusion complex in aqueous environment were performed. The starting set of






Phosphodiester models for cleavage of nucleic acids
Beilstein J. Org. Chem. 2018, 14, 803–837, doi:10.3762/bjoc.14.68

- ). Combined QM/MM simulations have lent support for this interpretation [47]. With triester analogs, such as uridine 3´-diethyl phosphate, the latter intramolecular proton transfer is not possible and the ratio kcl/kis is much smaller than with the diester analog, around 10−5 [50]. Since the barrier for the
























































Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids – tutorial
Beilstein J. Org. Chem. 2018, 14, 84–105, doi:10.3762/bjoc.14.5
















The use of 4,4,4-trifluorothreonine to stabilize extended peptide structures and mimic β-strands
Beilstein J. Org. Chem. 2017, 13, 2842–2853, doi:10.3762/bjoc.13.276

- dynamics simulations. CF3-threonine containing pentapeptides are more prone to mimic β-strands than their natural Ser and Thr pentapeptide analogues. The proof of concept that these fluorinated β-strand mimics are able to disrupt protein–protein interactions involving β-sheet structures is provided. The
- according to their central fluorinated or non-fluorinated residue, all-atom molecular dynamics (MD) simulations were performed using the GROMACS 4.5 package, with the OPLS-AA force field in combination with the SPC/E water model (for a complete description of the method, see Supporting Information File 1
- in order to better anticipate the peptide conformations in a solvent closer to physiological conditions. Nevertheless, we verified for compounds 2b and 4b that the simulations conducted in MeOH and in water were very similar (Figure S23, Supporting Information File 1). Overall, the theoretical 3JHN








What contributes to an effective mannose recognition domain?
Beilstein J. Org. Chem. 2017, 13, 2584–2595, doi:10.3762/bjoc.13.255

- well as the influence of highly mobile vs conserved waters were analyzed. For the assessment of the dynamic behavior of the ligand complexes of the seven calcium-dependent lectins, 20 ns molecular dynamics (MD) simulations were performed [57]. The most prominent interactions of O–C3 and O–C4 of the
- calcium ions. During MD simulations, the number of ligand–protein hydrogen-bond interactions for lectins A–F varied from 1.5 to 3.5, and subsequently increased to 4.5 and 5.4 for LecB (G) and BC2L-A (H), respectively. Lastly, FimH (I) forms on average 7.9 hydrogen bonds with methyl α-D-mannopyranoside (2
- close to the first calcium ion, a process which is hindered by His112 in H, leading to a 25-fold difference in affinity. However, in the case of highly mobile water molecules, water-mediated H-bonds as observed in MD simulations destabilize the carbohydrate–lectin interaction, whereas a pre-constrained









Homologated amino acids with three vicinal fluorines positioned along the backbone: development of a stereoselective synthesis
Beilstein J. Org. Chem. 2017, 13, 2316–2325, doi:10.3762/bjoc.13.228

- compounds. Model studies that informed the final steps of the synthesis. Supporting Information Supporting Information File 300: Synthetic procedures and characterisation data of intermediated, NMR spectra and NMR simulations for 6a,b. Acknowledgements L.H. thanks the Australian Research Council for







2-Methyl-2,4-pentanediol (MPD) boosts as detergent-substitute the performance of ß-barrel hybrid catalyst for phenylacetylene polymerization
Beilstein J. Org. Chem. 2017, 13, 1498–1506, doi:10.3762/bjoc.13.148

- rhodium-based biohybrid catalyst. Unlike commonly used detergents such as sodium dodecyl sulfate or polyethylene polyethyleneglycol, MPD does not form micelles in solution. Molecular dynamics simulations revealed the effect and position of stabilizing MPD molecules. The advantage of the amphiphilic MPD
- 1). Molecular dynamics (MD) simulations reveal an optimal minimum number of ≈200 MPD molecules for shielding the hydrophobic transmembrane region of FhuA ΔCVFtev MD simulations of FhuA ΔCVFtev were performed in a box with varying numbers of MPD molecules from 126 MPD, 189 MPD, 252 MPD to 378 MPD
- molecules as stabilizing cosolvent to investigate the molecular dynamics of protein structure stabilization, how a small amphiphilic molecule could stabilize a transmembrane protein such as FhuA ΔCVFtev. All simulations started with a random distribution of MPD, but after a few nanoseconds, the MPD






Grip on complexity in chemical reaction networks
Beilstein J. Org. Chem. 2017, 13, 1486–1497, doi:10.3762/bjoc.13.147

- were determined from kinetic studies in isolated individual reactions, allowing accurate simulations to test specific details of the experiments. We used the model to vary the rate constant that is induced by the changes to the molecular structure. First we show in Figure 7b that the tuning of R1
- predator–prey network. Adapted with permission from [88], copyright 2013 American Chemical Society. (b) The gene regulation pathway and of an oscillator based on a positive and delayed negative feedback motif, with experimentally observed oscillations shown to be in good agreement with the simulations
- . Adapted with permission from [90], copyright 2011 the authors. (c) Oscillations in the dual-feedback motif. (d) Illustration of the explicit intermediate processes required for accurate simulations in the mathematical modelling of genetic reaction networks. Adapted with permission from [91], copyright









Towards open-ended evolution in self-replicating molecular systems
Beilstein J. Org. Chem. 2017, 13, 1189–1203, doi:10.3762/bjoc.13.118

- uncatalyzed pathways and can therefore have a value smaller than 1, even for cases where the autocatalytic pathway itself would have a reaction order r = 1. In such situations computational simulations of the system can provide additional information on the replication processes that are involved [29]. 2.4














Kinetic analysis of mechanoradical formation during the mechanolysis of dextran and glycogen
Beilstein J. Org. Chem. 2017, 13, 1174–1183, doi:10.3762/bjoc.13.116

- on ESR spectra coupled with systematic computer simulations, in comparison with the mechanolysis of amylose. The component spectra of Dx and Gly were essentially identical to those of amylose and remained nearly unchanged in the course of vibratory milling. Simulated Dx, Gly, and amylose spectra were
- also obtained from admixtures of the component spectra at different ratios. Computer simulations revealed that a singlet spectrum (II) assignable to the immobilized DBS was a major component of milled Dx and Gly. The generated Dx and Gly mechanoradicals dissipated more readily than amylose
- particle diameter were determined by the histogram method with the Marquardt calculation. Computer simulations of ESR spectra Analogous to the description in [5], computational simulations were performed on a personal computer (DELL Inspiron 545S) using a simulation program developed in our laboratory. The











Correlation of surface pressure and hue of planarizable push–pull chromophores at the air/water interface
Beilstein J. Org. Chem. 2017, 13, 1099–1105, doi:10.3762/bjoc.13.109

- . Computational simulations Geometry optimization, as well as frequency calculations for the flipper mechanophore, were performed in the gas phase at the density functional level of theory with the Gaussian 03 program package [35] using the hybrid B3LYP functional [36] in conjunction with the LanL2DZ basis set






G-Protein coupled receptors: answers from simulations
Beilstein J. Org. Chem. 2017, 13, 1071–1078, doi:10.3762/bjoc.13.106

- Timothy Clark Computer-Chemie-Centrum, Department of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nuernberg, Naegelsbachstr. 25, 91052 Erlangen, Germany 10.3762/bjoc.13.106 Abstract Molecular-dynamics (MD) simulations are playing an increasingly important role in research into
- the modes of action of G-protein coupled receptors (GPCRs). In this field, MD simulations are unusually important as, because of the difficult experimental situation, they often offer the only opportunity to determine structural and mechanistic features in atomistic detail. Modern combinations of soft
- - and hardware have made MD simulations a powerful tool in GPCR research. This is important because GPCRs are targeted by approximately half of the drugs on the market, so that computer-aided drug design plays a major role in GPCR research. Keywords: computer-aided drug design; GPCR; metadynamicxs





Cycloheximide congeners produced by Streptomyces sp. SC0581 and photoinduced interconversion between (E)- and (Z)-2,3-dehydroanhydrocycloheximides
Beilstein J. Org. Chem. 2017, 13, 1039–1049, doi:10.3762/bjoc.13.103

- new compounds were achieved by spectroscopic analysis in combination with theoretical conformational analysis and ECD simulations, in which theoretical computations were shown to play a key role in solving challenges in assignments of relative and absolute configurations. Analysis of the antifungal







Aggregation behaviour of a single-chain, phenylene-modified bolalipid and its miscibility with classical phospholipids
Beilstein J. Org. Chem. 2017, 13, 995–1007, doi:10.3762/bjoc.13.99

- simulations [27]. A temperature increase leads to a transformation of the nanofibres into small micelles and the gel character is lost. This reversible gel/sol transformation is accompanied by a cooperative endothermic transition at Tm = 48 °C, which can be followed by differential scanning calorimetry (DSC










Automating multistep flow synthesis: approach and challenges in integrating chemistry, machines and logic
Beilstein J. Org. Chem. 2017, 13, 960–987, doi:10.3762/bjoc.13.97

- (viz. forcefully quenching the reaction). There should also be a sequence of operation procedures which can take the process from one state of operating conditions to another state of operation. Dynamic simulations can be a useful tool to study the special purpose operations (viz. start-up, shut-down



















Membrane properties of hydroxycholesterols related to the brain cholesterol metabolism
Beilstein J. Org. Chem. 2017, 13, 720–727, doi:10.3762/bjoc.13.71

- fluorescence techniques [21]. Also, a decreased but still significant effect of the hydroxysterols on lipid condensation compared to native cholesterol was found in molecular dynamics simulations, which is probably caused by an increased tilt angle of the sterols to the membrane normal [8][21]. However, using
- estradiol was found in the lipid water interface of the membrane with an orientation perpendicular to the membrane normal. By that, the molecules act disturbing rather than ordering. Notably, MD simulations found an opposite effect of the hydroxycholesterols [21]. The additional polarity of
- proteins) is impacted. MD simulations for 27-HC have shown, that this molecule adopts compared to cholesterol different orientations within the membrane, which are upside-down, largely tilted and/or inter-leaflet positions [21]. These properties indicate that the molecules are less strongly anchored within




