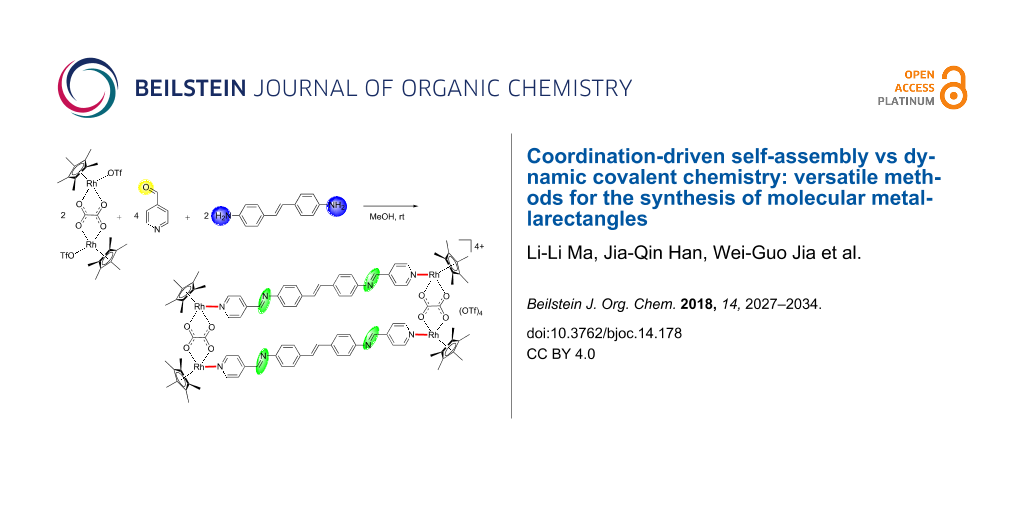
Abstract
Supramolecular coordination assemblies have a range of potential applications in chemical and biological sciences. Herein, simple modular methods for the synthesis of metallarectangles are described. The desired tetranuclear metallarectangles were synthesized by using coordination-driven self-assembly of half-sandwich rhodium-based organometallic clip units and organic ligands. The reaction of such an organometallic clip with 4-formylpyridine provided a dinuclear molecular tweezer with pendant aldehyde groups, and subsequent [4 + 4] condensation reactions with diamines provides another route to the target metallarectangles in good yields. The same assemblies can also be easily isolated in one-pot procedures by mixing the organometallic clip, diamines and 4-formylpyridine.

Graphical Abstract
Introduction
Over the past two decades, supramolecular structures with organometallic half-sandwich fragments have attracted much attention, including metallarectangles, metallacages and Borromean-type rings. Moreover, many of these structures have been utilized for various applications, such as catalysts, host–guest chemistry and others [1-17]. Through the use of a range of diverse functional ligands, the coordination-driven self-assembly has been proven to be a powerful tool to construct supramolecular architectures with controlled shapes and sizes [18-30]. Using this strategy, a host of exciting supramolecular structures have been constructed by using two elaborately designed building blocks, such as dinuclear half-sandwich molecular clips and appropriate pyridyl ligands. The sizes and structures of the obtained molecular rectangles, cages or rings can be easily tuned by adjusting the length and shape of the bridging ligands and molecular clips. We and others have reported a suite of [2 + 2] tetranuclear metallarectangles, each formed using dinuclear molecular clips and pyridyl-based donor ligands [6-9,31-36]. The introduction of dynamic covalent bonds (such as imine C=N bonds), could allow the multicomponent assembly of such architectures using rather simple precursors, however, studies along these lines are rare [37]. Thus, the preparation of single and discrete supramolecular architectures via dynamic covalent bond-driven self-assembly remains challenging. Severin and co-workers have recently shown that metallamacrocycles and cages based on half-sandwich ruthenium could be obtained in one-pot reactions from simple building blocks [38,39]. This finding prompted us to investigate whether condensation reactions between amines and 4-formylpyridine can be used simultaneously with coordination bond formation to construct metallarectangle structures in one-pot reactions, thereby reducing both waste and the number of reaction steps.
In this work we successfully combine coordination-driven self-assembly and dynamic covalent chemistry through imine bond formation between amines and 4-formylpyridine to construct the desired rectangular tetrarhodium molecular rectangles.
Results and Discussion
The different approaches to the synthesis of tetranuclear molecular rectangles used in this work are shown in Scheme 1. We and others have used a two-step supramolecular design strategy for the formation of half-sandwich metal-based metallarectangles and metallacages [6-9]. Following this approach, two self-assembled metallarectangles with different bridging linkers 3a,b were synthesized by utilizing the [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] unit as molecular clips (Scheme 1, method A).
![[1860-5397-14-178-i1]](/bjoc/content/inline/1860-5397-14-178-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of half-sandwich rhodium metallarectangles via three different methods. Method A: coordination-driven self-assembly of organometallic clips and organic ligands; method B: [4 + 4] condensation reactions of half-sandwich rhodium-based dialdehyde complexes with diamines; method C: assembly of metallarectangles with organometallic clip, diamines and 4-formylpyridine in a one-pot procedure.
Scheme 1: Synthesis of half-sandwich rhodium metallarectangles via three different methods. Method A: coordin...
Precursor complex 1, which bears two labile triflato ligands was prepared in situ by chloride abstraction from [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] with AgOTf. Stirring a mixture of 1 and L1 in a 1:1 molar ratio in methanol for 24 h resulted in a homogeneous, dark-red solution. The 1H NMR spectrum of the obtained solution displays significant downfield shifts of the pyridyl signals, consistent with the loss of electron density upon coordination of the nitrogen atom to the metal centers (Figure 1a,b). Analysis of the reaction solution using electrospray ionization mass spectrometry (ESIMS) showed a signal at m/z = 476.1010, corresponding to a tertracation species of complex 3a. The peak was isotopically resolved and agrees well with the theoretical isotopic distribution. In addition, the IR spectrum of the rhodium metallarectangle 3a showed a C=N stretching band at 1618 cm−1.
![[1860-5397-14-178-1]](/bjoc/content/figures/1860-5397-14-178-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Partial 1H NMR spectra (400 MHz, DMSO-d6, ppm) of (a) L1; (b) the sample of metallarectagle 3a obtained by coordination-driven self-assembly of organometallic clip 1 and L1 (method A); (c) the sample of metallarectagle 3a obtained through [4 + 4] condensation reactions of half-sandwich rhodium-based dialdehyde complex 2 with trans-4,4'-stilbenediamine (method B); (d) half-sandwich rhodium-based dialdehyde 2; (e) the product of self-assembly of organometallic clip 1, trans-4,4'-stilbenediamine and 4-formylpyridine in a one-pot procedure (method C).
Figure 1: Partial 1H NMR spectra (400 MHz, DMSO-d6, ppm) of (a) L1; (b) the sample of metallarectagle 3a obta...
The same self-assembly protocol can also be used for the synthesis of metallarectangle 3b. The combination of two labile-ligand precursor complexes 1 and two pyridyl-based ligands L2 in a 1:1 molar ratio led to the formation of 3b in good yield. The 1H NMR spectrum of the reaction mixture revealed the formation of a single species. In the 1H NMR spectrum of 3b, only one sharp set of characteristic peaks was found. Again, significant downfield shifts of the pyridyl proton signals were observed, indicating the efficient self-assembly of the rhodium-based assembly (Figure 2a,b). Clear evidence for the formation of a discrete tetranuclear organometallic product was obtained from ESI mass spectrometry. Similar to that observed in complex 3a, a peak at m/z = 450.0868 was observed, which is attributable to [3b − 4OTf]4+, and its isotopic pattern is in good agreement with the theoretical distribution (Figure 3, right). The absorption band at 1620 cm−1 in the IR spectrum indicated the existence of an imine group.
![[1860-5397-14-178-2]](/bjoc/content/figures/1860-5397-14-178-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Partial 1H NMR spectra (400 MHz, DMSO-d6, ppm) of (a) L2; (b) a sample of metallarectangle 3b obtained by coordination-driven self-assembly of organometallic clip 1 and L2 (method A); (c) a sample of metallarectangle 3b obtained by [4 + 4] condensation reactions of half-sandwich rhodium-based dialdehyde complex 2 with 1,5-diaminonaphthalene (method B); (d) half-sandwich rhodium-based dialdehyde 2; (e) the product of assembly of organometallic clip 1, 1,5-diaminonaphthalene and 4-formylpyridine in a one-pot procedure (method C).
Figure 2: Partial 1H NMR spectra (400 MHz, DMSO-d6, ppm) of (a) L2; (b) a sample of metallarectangle 3b obtai...
![[1860-5397-14-178-3]](/bjoc/content/figures/1860-5397-14-178-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Calculated (bottom) and experimental (top) ESI-MS spectra of the tetracationic half-sandwich rhodium metallarectangles [3a – 4OTf]4+ (left) and [3b – 4OTf]4+ (right).
Figure 3: Calculated (bottom) and experimental (top) ESI-MS spectra of the tetracationic half-sandwich rhodiu...
The geometries of the metallarectangles 3a and 3b were expected to be similar, as they comprise two oxalate-bridged half-sandwich rhodium fragments linked by two Schiff-base ligands L1 or L2, giving the desired tetranuclear metallarectangles. In order to test the possibility of using dynamic covalent chemistry to assemble these metallarectangles, we attempted a further method (Scheme 1, method B) to synthesize these assemblies. As shown in Scheme 1, a dinuclear molecular tweezer complex 2 bearing two pendant aldehyde groups can be formed from the labile ligand complex 1 and 4-formylpyridine, and subsequent reaction with diamines would potentially give tetranuclear metallarectangles. When equimolar amounts of either trans-4,4'-stilbenediamine or 1,5-diaminonaphthalene were added to methanol solutions of complex 2, and allowed to react for 24 h at room temperature, the formation of tetranuclear [4 + 4] condensation products 3a (Figure 1c) and 3b (Figure 2c) was observed, respectively. Complexes 3a and 3b were isolated in good yields, and their structures were confirmed by 1H NMR spectroscopy and ESI mass spectrometry.
After establishing that the condensation reaction of 2 with amines is an efficient method to form metallarectangles, we sought to test the possibility of forming the desired assemblies in a one-pot reaction, i.e., the combination of coordination-driven and dynamic covalent self-assembly strategies (Scheme 1, method C) [33]. When a mixture of the labile ligand complex 1, trans-4,4'-stilbenediamine and 4-formylpyridine in a 1:1:1 molar ratio in methanol was allowed to react for 24 h at room temperature, the clear, quantitative formation of complex 3a was revealed by NMR spectrometry (Figure 1e). The analogous one-pot construction of 3b was also successful (Figure 2e). Notably, the isolated yields of the metallarectangles are higher than the overall yields of the two-step method.
Since attempts to obtain X-ray quality single crystals of the target metallarectangles were unsuccessful, molecular simulations were performed to gain further insight into the structures of the assemblies 3a and 3b. The optimized structures of each assembly featured a similar rectangular metallacyclic macrocycle structure (Figure 4). The sizes of the assembled structures were estimated to be 26.3 × 5.6 Å (3a) and 20.1 × 5.6 Å (3b).
![[1860-5397-14-178-4]](/bjoc/content/figures/1860-5397-14-178-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Optimized structures of the charged metallarectangles 3a (top) and 3b (bottom), optimized with the molecular mechanics force field. The graphics were produced using the Diamond software package. Colors: C, gray; O, red; N, blue; rhodium, green (hydrogen atoms have been removed for clarity).
Figure 4: Optimized structures of the charged metallarectangles 3a (top) and 3b (bottom), optimized with the ...
Conclusion
In summary, a modular protocol for the synthesis of metallarectangles is described. The desired tetranuclear metallarectangles can be obtained via three different approaches: 1) exploiting the coordination-driven self-assembly of half-sandwich rhodium-based organometallic clips and organic ligands, 2) [4 + 4] condensation reactions of diamines with dinuclear molecular tweezer complex bearing pendant aldehyde groups, and 3) a sample one-pot procedure involving mixing the organometallic clips, diamines and 4-formylpyridine. Our results thus present versatile and efficient approaches to the synthesis of molecular metallarectangles with intricate topologies. The methods shown here are potentially useful for the synthesis of functional molecular metallacages, and the experimental efforts in this direction are currently underway.
Experimental
Materials and methods
All manipulations were performed under an atmosphere of nitrogen using standard Schlenk techniques. Commercial grade solvents and reagents were used without further purification. [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] [25], trans-4,4'-stilbenediamine [40] and L2 [41] were prepared according to literature procedures. Methanol was purified by standard methods prior to use. NMR (400 MHz) spectra were obtained on a Bruker AVANCE III spectrometer. IR spectra of the solid samples (KBr tablets) were recorded on a Bruker EQUINOX-55 (TENSOR27) IR spectrometer. Mass spectra were obtained with UltiMate3000 spectrometers.
General procedure for the synthesis of L1 and L2
L1: To 4-formylpyridine (195 mg, 1.82 mmol) in dry CH3OH (20 mL) was added trans-4,4'-stilbenediamine (191 mg, 0.91 mmol) at room temperature. The mixture was stirred at room temperature for 24 h and then filtered. The resulting yellow solid was washed with methanol (2 × 5 mL) and diethyl ether (2 × 5 mL) to give L1 (318 mg, 90%). 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.77 (s, 2H, -NCH-), 8.76 (d, J = 6.0 Hz, 4H), 7.87 (d, J = 6.0 Hz, 4H), 7.72 (d, J = 8.4 Hz, 4H), 7.41 (d, J = 8.4 Hz, 4H), 7.36 (s, 2H, -CH=CH-); IR (KBr, cm−1): 3419 (m), 1597 (s), 1411 (m), 962 (m), 831 (s), 632 (m), 561 (s).
L2: A modified synthetic procedure adapted from literature methods [33] was used. To 4-formylpyridine (215 mg, 2.0 mmol) in dry CH3OH (20 mL) was added 1,5-diaminonaphthalene (160 mg, 1.0 mmol) at room temperature. The mixture was stirred at room temperature for 24 h and then filtered. The resulting yellow solid was washed with diethyl ether (2 × 3 mL) and crystallized from CH2Cl2/hexane (1:1) to give L2 (220 mg, 65%). 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.83 (s, 2H, -NCH-), 8.82 (d, J = 6.0 Hz, 4H), 8.26 (d, J = 8.4 Hz, 2H), 8.01 (d, J = 6.0 Hz, 4H), 7.62 (t, 2H), 7.40 (d, J = 7.2 Hz, 2H); IR (KBr, cm−1): 3024 (w), 1624 (s), 1597 (s), 1404 (s), 1317 (s), 1232 (s), 925 (s), 790 (s), 652 (m).
Synthesis of complex 2 [Cp*2Rh2(μ-η2-η2-C2O4)(4-CHOPy)2](OTf)2
AgOTf (36 mg, 0.14 mmol) was added to a solution of [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] (45 mg, 0.07 mmol) in CH3OH (15 mL) at room temperature and the mixture was stirred for 1 h, followed by filtration to remove insoluble materials. Then 4-formylpyridine (16 mg, 0.14 mmol) was added to the filtrate and the mixture was stirred for 24 h. The volume was reduced to 3 mL in vacuo. Upon the addition of diethyl ether, a light-yellow solid was precipitated and washed with diethyl ether (3 × 3 mL) and dried under vacuum (60 mg, 80%).1H NMR (400 MHz, DMSO-d6, ppm) δ 10.09 (s, 2H, -CHO), 8.89 (d, J = 5.0 Hz, 4H), 7.82 (d, J = 5.0 Hz, 4H), 1.55 (s, 30H, Cp*-H); HRMS–ESI (m/z): [2 − 2OTf]2+ calcd for C34H40O12N2F6S2Rh2, 390.0571; found, 390.0541.
Synthesis of [Cp*4Rh4(μ-η2-η2-C2O4)2(L1)2](OTf)4 (3a)
Method A: AgOTf (36 mg, 0.14 mmol) was added to a solution of [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] (45 mg, 0.07 mmol) in CH3OH (30 mL) at room temperature and the mixture was stirred for 1 h, followed by filtration to remove insoluble materials. Then a solution of L1 (27 mg, 0.07 mmol) in 15 mL CHCl3 was added dropwise to the filtrate. The mixture was stirred at room temperature for 24 h to give a deep red solution. The volume was reduced to 3 mL in vacuo. Upon the addition of diethyl ether, a black-red solid was precipitated and washed with CHCl3 (2 × 3 mL) and dried under vacuum (61 mg, 70%). 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.64 (s, 4H, -NCH-), 8.18 (d, J = 6.0 Hz, 8H), 7.81 (d, J = 6.4 Hz, 8H), 7.36 (d, J = 8.8 Hz, 8H), 7.12 (d, J = 6.0 Hz, 8H), 7.09 (s, 4H), 1.57 (s, 60H, Cp*-H). HRMS–ESI (m/z): [3a − 4OTf]4+ calcd for C100H100O20N8F12S4Rh4, 476.0966; found, 476.0986.
Method B: trans-4,4'-Stilbenediamine (14 mg, 0.07 mmol) was added to a solution of 2 (76 mg, 0.07 mmol) in CH3OH (15 mL) at room temperature and the mixture was stirred for 24 h to give a deep red solution. Upon the addition of diethyl ether, a black-red solid was precipitated and washed with diethyl ether (3 × 3 mL) and dried under vacuum (63 mg, 72%). HRMS–ESI (m/z): [3a − 4OTf]4+ calcd for C100H100O20N8F12S4Rh4, 476.0966; found, 476.0963.
Method C: AgOTf (36 mg, 0.14 mmol) was added to a solution of [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] (45 mg, 0.07 mmol) in CH3OH (10 mL) at room temperature and the mixture was stirred for 1 h, followed by filtration to remove insoluble materials. Then trans-4,4'-stilbenediamine (15 mg, 0.07 mmol) was added to the filtrate. A solution of 4-formylpyridine (15 mg, 0.14 mmol) in 7 mL CHCl3 was then added dropwise to the mixture and stirred for 24 h. The solvent was concentrated to about 3 mL. Diethyl ether was then added, and a black-red solid precipitated, which was washed with diethyl ether (3 × 3 mL) and chloroform (2 × 3 mL) and dried under vacuum (66 mg, 75%). HRMS–ESI (m/z): [3a − 4OTf]4+ calcd for C100H100O20N8F12S4Rh4, 476.0966; found, 476.1010.
Synthesis of [Cp*4Rh4(μ-η2-η2-C2O4)2(L2)2](OTf)4 (3b)
Method A: AgOTf (36 mg, 0.14 mmol) was added to a solution of [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] (45 mg, 0.07 mmol) in CH3OH (15 mL) at room temperature and the mixture was stirred for 1 h, followed by filtration to remove insoluble materials. Then a solution of L2 (24 mg, 0.07 mmol) in 7 mL CHCl3 was added dropwise to the filtrate. The mixture was stirred at room temperature for 24 h and filtered. The resulting yellow solid was washed with chloroform (2 × 3 mL) and dried under vacuum (61 mg, 70%). 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.74 (s, 4H, -NCH-), 8.25 (d, J = 6.0 Hz, 8H), 7.98 (d, J = 6.0 Hz, 8H), 7.87 (d, J = 8.8 Hz, 4H), 7.13 (t, 4H), 7.05 (d, J = 6.8 Hz, 4H), 1.59 (s, 60H, Cp*-H); HRMS–ESI (m/z): [3b − 4OTf]4+ calcd for C92H92O20N8F12S4Rh4, 450.0809; found, 450.0873.
Method B: 1,5-Diaminonaphthalene (11 mg, 0.07 mmol) was added to a solution of 2 (76 mg, 0.07 mmol) in CH3OH (20 mL) at room temperature and the mixture was stirred for 24 h to give a deep red solution. The volume was reduced to 3 mL in vacuo. Upon addition of diethyl ether, a yellow solid precipitated, which was washed with diethyl ether (3 × 5 mL) and dried under vacuum (61 mg, 73%). HRMS–ESI (m/z): [3b − 4OTf]4+ calcd for C92H92O20N8F12S4Rh4, 450.0809; found, 450.0807.
Method C: AgOTf (36 mg, 0.14 mmol) was added to a solution of [Cp*2Rh2(μ-η2-η2-C2O4)Cl2] (45 mg, 0.07 mmol) in CH3OH (15 mL) at room temperature and the mixture was stirred for 1 h, followed by filtration to remove insoluble materials. Then, 1,5-diaminonaphthalene (11 mg, 0.07mmol) was added to the filtrate. A solution of 4-formylpyridine (15 mg, 0.14 mmol) in 7 mL CHCl3 was added dropwise to the mixture and stirred for 24 h. A yellow solid precipitated, which was washed with chloroform (2 × 3 mL) and dried under vacuum (60 mg, 72%). HRMS–ESI (m/z): [3b − 4OTf]4+ calcd for C92H92O20N8F12S4Rh4, 450.0809; found, 450.0868.
Acknowledgements
The authors gratefully acknowledge financial support from the NSFC (Nos. 21722105, 21102004, 21771146), the Scientific Research Foundation for the Returned Overseas Scholars of Shaanxi Province (2017001), the Shaanxi Key Laboratory of Physical-inorganic Chemistry (17JS133), the FM & EM International Joint Laboratory of Northwest University, and NSF of Anhui Province (1708085MB44).
References
-
Fish, R. H. Coord. Chem. Rev. 1999, 185–186, 569–584. doi:10.1016/S0010-8545(99)00011-9
Return to citation in text: [1] -
Severin, K. Coord. Chem. Rev. 2003, 245, 3–10. doi:10.1016/S0010-8545(03)00028-6
Return to citation in text: [1] -
Severin, K. Chem. Commun. 2006, 3859–3867. doi:10.1039/b606632c
Return to citation in text: [1] -
Boyer, J. L.; Kuhlman, M. L.; Rauchfuss, T. B. Acc. Chem. Res. 2007, 40, 233–242. doi:10.1021/ar050215j
Return to citation in text: [1] -
Thanasekaran, P.; Lee, C.-C.; Lu, K.-L. Acc. Chem. Res. 2012, 45, 1403–1418. doi:10.1021/ar200243w
Return to citation in text: [1] -
Cook, T. R.; Vajpayee, V.; Lee, M. H.; Stang, P. J.; Chi, K.-W. Acc. Chem. Res. 2013, 46, 2464–2474. doi:10.1021/ar400010v
Return to citation in text: [1] [2] [3] -
Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j
Return to citation in text: [1] [2] [3] -
Han, Y.-F.; Jin, G.-X. Chem. Soc. Rev. 2014, 43, 2799–2823. doi:10.1039/C3CS60343A
Return to citation in text: [1] [2] [3] -
Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571–3579. doi:10.1021/ar500335a
Return to citation in text: [1] [2] [3] -
Lu, Y.; Deng, Y.-X.; Lin, Y.-J.; Han, Y.-F.; Weng, L.-H.; Li, Z.-H.; Jin, G.-X. Chemistry 2017, 3, 110–121. doi:10.1016/j.chempr.2017.06.006
Return to citation in text: [1] -
Mirtschin, S.; Slabon-Turski, A.; Scopelliti, R.; Velders, A. H.; Severin, K. J. Am. Chem. Soc. 2010, 132, 14004–14005. doi:10.1021/ja1063789
Return to citation in text: [1] -
Conrady, F. M.; Fröhlich, R.; Schulte to Brinke, C.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2011, 133, 11496–11499. doi:10.1021/ja205021p
Return to citation in text: [1] -
Lee, H.; Elumalai, P.; Singh, N.; Kim, H.; Lee, S. U.; Chi, K.-W. J. Am. Chem. Soc. 2015, 137, 4674–4677. doi:10.1021/jacs.5b02573
Return to citation in text: [1] -
Zhang, W.-Y.; Lin, Y.-J.; Han, Y.-F.; Jin, G.-X. J. Am. Chem. Soc. 2016, 138, 10700–10707. doi:10.1021/jacs.6b06622
Return to citation in text: [1] -
Liu, Q.; Zhang, W.-H.; Lang, J.-P. Coord. Chem. Rev. 2017, 350, 248–274. doi:10.1016/j.ccr.2017.06.027
Return to citation in text: [1] -
Lang, J.-P.; Xu, Q.-F.; Chen, Z.-N.; Abrahams, B. F. J. Am. Chem. Soc. 2003, 125, 12682–12683. doi:10.1021/ja036995d
Return to citation in text: [1] -
Zhang, W.-H.; Ren, Z.-G.; Lang, J.-P. Chem. Soc. Rev. 2016, 45, 4995–5019. doi:10.1039/C6CS00096G
Return to citation in text: [1] -
Wang, W.; Wang, Y.-X.; Yang, H.-B. Chem. Soc. Rev. 2016, 45, 2656–2693. doi:10.1039/C5CS00301F
Return to citation in text: [1] -
Cotton, F. A.; Lin, C.; Murillo, C. A. Acc. Chem. Res. 2001, 34, 759–771. doi:10.1021/ar010062+
Return to citation in text: [1] -
Fujita, M.; Tominaga, M.; Hori, A.; Therrien, B. Acc. Chem. Res. 2005, 38, 369–378. doi:10.1021/ar040153h
Return to citation in text: [1] -
Caulder, D. L.; Raymond, K. N. Acc. Chem. Res. 1999, 32, 975–982. doi:10.1021/ar970224v
Return to citation in text: [1] -
Saalfrank, R. W.; Maid, H.; Scheurer, A. Angew. Chem., Int. Ed. 2008, 47, 8794–8824. doi:10.1002/anie.200702075
Return to citation in text: [1] -
Qiu, X.-T.; Yao, R.; Zhou, W.-F.; Liu, M.-D.; Liu, Q.; Song, Y.-L.; Young, D. J.; Zhang, W.-H.; Lang, J.-P. Chem. Commun. 2018, 54, 4168–4171. doi:10.1039/C8CC01950A
Return to citation in text: [1] -
Zhang, W.-H.; Liu, Q.; Lang, J.-P. Coord. Chem. Rev. 2015, 293–294, 187–210. doi:10.1016/j.ccr.2014.12.010
Return to citation in text: [1] -
Lang, J.-P.; Xu, Q.-F.; Yuan, R.-X.; Abrahams, B. F. Angew. Chem., Int. Ed. 2004, 43, 4741–4745. doi:10.1002/anie.200460076
Return to citation in text: [1] [2] -
Liu, D.; Lang, J.-P.; Abrahams, B. F. J. Am. Chem. Soc. 2011, 133, 11042–11045. doi:10.1021/ja203053y
Return to citation in text: [1] -
Sun, L.-Y.; Sinha, N.; Yan, T.; Wang, Y.-S.; Tan, T. T. Y.; Yu, L.; Han, Y.-F.; Hahn, F. E. Angew. Chem., Int. Ed. 2018, 57, 5161–5165. doi:10.1002/anie.201713240
Return to citation in text: [1] -
Cook, T. R.; Stang, P. J. Chem. Rev. 2015, 115, 7001–7045. doi:10.1021/cr5005666
Return to citation in text: [1] -
Zhang, Y.-Y.; Gao, W.-X.; Lin, L.; Jin, G.-X. Coord. Chem. Rev. 2017, 344, 323–344. doi:10.1016/j.ccr.2016.09.010
Return to citation in text: [1] -
Wu, G.-Y.; Chen, L.-J.; Xu, L.; Zhao, X.-L.; Yang, H.-B. Coord. Chem. Rev. 2018, 369, 39–75. doi:10.1016/j.ccr.2018.05.009
Return to citation in text: [1] -
Zhang, H.-N.; Gao, W.-X.; Deng, Y.-X.; Lin, Y.-J.; Jin, G.-X. Chem. Commun. 2018, 54, 1559–1562. doi:10.1039/C7CC09448E
Return to citation in text: [1] -
Han, Y.-F.; Li, H.; Jin, G.-X. Chem. Commun. 2010, 46, 6879–6890. doi:10.1039/c0cc00770f
Return to citation in text: [1] -
Han, Y.-F.; Lin, Y.-J.; Jia, W.-G.; Jin, G.-X. Organometallics 2008, 27, 4088–4097. doi:10.1021/om800426e
Return to citation in text: [1] [2] [3] -
Han, Y.-F.; Jia, W.-G.; Lin, Y.-J.; Jin, G.-X. Organometallics 2008, 27, 5002–5008. doi:10.1021/om800490s
Return to citation in text: [1] -
Han, Y.-F.; Jia, W.-G.; Lin, Y.-J.; Jin, G.-X. Angew. Chem., Int. Ed. 2009, 48, 6234–6238. doi:10.1002/anie.200805949
Return to citation in text: [1] -
Vajpayee, V.; Bivaud, S.; Goeb, S.; Croué, V.; Allain, M.; Popp, B. V.; Garci, A.; Therrien, B.; Sallé, M. Organometallics 2014, 33, 1651–1658. doi:10.1021/om401142j
Return to citation in text: [1] -
Zarra, S.; Wood, D. M.; Roberts, D. A.; Nitschke, J. R. Chem. Soc. Rev. 2015, 44, 419–432. doi:10.1039/C4CS00165F
Return to citation in text: [1] -
Granzhan, A.; Schouwey, C.; Riis-Johannessen, T.; Scopelliti, R.; Severin, K. J. Am. Chem. Soc. 2011, 133, 7106–7115. doi:10.1021/ja200580x
Return to citation in text: [1] -
Schouwey, C.; Scopelliti, R.; Severin, K. Chem. – Eur. J. 2013, 19, 6274–6281. doi:10.1002/chem.201300098
Return to citation in text: [1] -
Liu, X.; Liu, H.; Zhou, W.; Zheng, H.; Yin, X.; Li, Y.; Guo, Y.; Zhu, M.; Ouyang, C.; Zhu, D.; Xia, A. Langmuir 2010, 26, 3179–3185. doi:10.1021/la903838w
Return to citation in text: [1] -
Min, D.; Cho, B.-Y.; Lee, S. W. Inorg. Chim. Acta 2006, 359, 577–584. doi:10.1016/j.ica.2005.09.041
Return to citation in text: [1]
| 1. | Fish, R. H. Coord. Chem. Rev. 1999, 185–186, 569–584. doi:10.1016/S0010-8545(99)00011-9 |
| 2. | Severin, K. Coord. Chem. Rev. 2003, 245, 3–10. doi:10.1016/S0010-8545(03)00028-6 |
| 3. | Severin, K. Chem. Commun. 2006, 3859–3867. doi:10.1039/b606632c |
| 4. | Boyer, J. L.; Kuhlman, M. L.; Rauchfuss, T. B. Acc. Chem. Res. 2007, 40, 233–242. doi:10.1021/ar050215j |
| 5. | Thanasekaran, P.; Lee, C.-C.; Lu, K.-L. Acc. Chem. Res. 2012, 45, 1403–1418. doi:10.1021/ar200243w |
| 6. | Cook, T. R.; Vajpayee, V.; Lee, M. H.; Stang, P. J.; Chi, K.-W. Acc. Chem. Res. 2013, 46, 2464–2474. doi:10.1021/ar400010v |
| 7. | Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j |
| 8. | Han, Y.-F.; Jin, G.-X. Chem. Soc. Rev. 2014, 43, 2799–2823. doi:10.1039/C3CS60343A |
| 9. | Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571–3579. doi:10.1021/ar500335a |
| 10. | Lu, Y.; Deng, Y.-X.; Lin, Y.-J.; Han, Y.-F.; Weng, L.-H.; Li, Z.-H.; Jin, G.-X. Chemistry 2017, 3, 110–121. doi:10.1016/j.chempr.2017.06.006 |
| 11. | Mirtschin, S.; Slabon-Turski, A.; Scopelliti, R.; Velders, A. H.; Severin, K. J. Am. Chem. Soc. 2010, 132, 14004–14005. doi:10.1021/ja1063789 |
| 12. | Conrady, F. M.; Fröhlich, R.; Schulte to Brinke, C.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2011, 133, 11496–11499. doi:10.1021/ja205021p |
| 13. | Lee, H.; Elumalai, P.; Singh, N.; Kim, H.; Lee, S. U.; Chi, K.-W. J. Am. Chem. Soc. 2015, 137, 4674–4677. doi:10.1021/jacs.5b02573 |
| 14. | Zhang, W.-Y.; Lin, Y.-J.; Han, Y.-F.; Jin, G.-X. J. Am. Chem. Soc. 2016, 138, 10700–10707. doi:10.1021/jacs.6b06622 |
| 15. | Liu, Q.; Zhang, W.-H.; Lang, J.-P. Coord. Chem. Rev. 2017, 350, 248–274. doi:10.1016/j.ccr.2017.06.027 |
| 16. | Lang, J.-P.; Xu, Q.-F.; Chen, Z.-N.; Abrahams, B. F. J. Am. Chem. Soc. 2003, 125, 12682–12683. doi:10.1021/ja036995d |
| 17. | Zhang, W.-H.; Ren, Z.-G.; Lang, J.-P. Chem. Soc. Rev. 2016, 45, 4995–5019. doi:10.1039/C6CS00096G |
| 38. | Granzhan, A.; Schouwey, C.; Riis-Johannessen, T.; Scopelliti, R.; Severin, K. J. Am. Chem. Soc. 2011, 133, 7106–7115. doi:10.1021/ja200580x |
| 39. | Schouwey, C.; Scopelliti, R.; Severin, K. Chem. – Eur. J. 2013, 19, 6274–6281. doi:10.1002/chem.201300098 |
| 37. | Zarra, S.; Wood, D. M.; Roberts, D. A.; Nitschke, J. R. Chem. Soc. Rev. 2015, 44, 419–432. doi:10.1039/C4CS00165F |
| 6. | Cook, T. R.; Vajpayee, V.; Lee, M. H.; Stang, P. J.; Chi, K.-W. Acc. Chem. Res. 2013, 46, 2464–2474. doi:10.1021/ar400010v |
| 7. | Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j |
| 8. | Han, Y.-F.; Jin, G.-X. Chem. Soc. Rev. 2014, 43, 2799–2823. doi:10.1039/C3CS60343A |
| 9. | Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571–3579. doi:10.1021/ar500335a |
| 31. | Zhang, H.-N.; Gao, W.-X.; Deng, Y.-X.; Lin, Y.-J.; Jin, G.-X. Chem. Commun. 2018, 54, 1559–1562. doi:10.1039/C7CC09448E |
| 32. | Han, Y.-F.; Li, H.; Jin, G.-X. Chem. Commun. 2010, 46, 6879–6890. doi:10.1039/c0cc00770f |
| 33. | Han, Y.-F.; Lin, Y.-J.; Jia, W.-G.; Jin, G.-X. Organometallics 2008, 27, 4088–4097. doi:10.1021/om800426e |
| 34. | Han, Y.-F.; Jia, W.-G.; Lin, Y.-J.; Jin, G.-X. Organometallics 2008, 27, 5002–5008. doi:10.1021/om800490s |
| 35. | Han, Y.-F.; Jia, W.-G.; Lin, Y.-J.; Jin, G.-X. Angew. Chem., Int. Ed. 2009, 48, 6234–6238. doi:10.1002/anie.200805949 |
| 36. | Vajpayee, V.; Bivaud, S.; Goeb, S.; Croué, V.; Allain, M.; Popp, B. V.; Garci, A.; Therrien, B.; Sallé, M. Organometallics 2014, 33, 1651–1658. doi:10.1021/om401142j |
| 18. | Wang, W.; Wang, Y.-X.; Yang, H.-B. Chem. Soc. Rev. 2016, 45, 2656–2693. doi:10.1039/C5CS00301F |
| 19. | Cotton, F. A.; Lin, C.; Murillo, C. A. Acc. Chem. Res. 2001, 34, 759–771. doi:10.1021/ar010062+ |
| 20. | Fujita, M.; Tominaga, M.; Hori, A.; Therrien, B. Acc. Chem. Res. 2005, 38, 369–378. doi:10.1021/ar040153h |
| 21. | Caulder, D. L.; Raymond, K. N. Acc. Chem. Res. 1999, 32, 975–982. doi:10.1021/ar970224v |
| 22. | Saalfrank, R. W.; Maid, H.; Scheurer, A. Angew. Chem., Int. Ed. 2008, 47, 8794–8824. doi:10.1002/anie.200702075 |
| 23. | Qiu, X.-T.; Yao, R.; Zhou, W.-F.; Liu, M.-D.; Liu, Q.; Song, Y.-L.; Young, D. J.; Zhang, W.-H.; Lang, J.-P. Chem. Commun. 2018, 54, 4168–4171. doi:10.1039/C8CC01950A |
| 24. | Zhang, W.-H.; Liu, Q.; Lang, J.-P. Coord. Chem. Rev. 2015, 293–294, 187–210. doi:10.1016/j.ccr.2014.12.010 |
| 25. | Lang, J.-P.; Xu, Q.-F.; Yuan, R.-X.; Abrahams, B. F. Angew. Chem., Int. Ed. 2004, 43, 4741–4745. doi:10.1002/anie.200460076 |
| 26. | Liu, D.; Lang, J.-P.; Abrahams, B. F. J. Am. Chem. Soc. 2011, 133, 11042–11045. doi:10.1021/ja203053y |
| 27. | Sun, L.-Y.; Sinha, N.; Yan, T.; Wang, Y.-S.; Tan, T. T. Y.; Yu, L.; Han, Y.-F.; Hahn, F. E. Angew. Chem., Int. Ed. 2018, 57, 5161–5165. doi:10.1002/anie.201713240 |
| 28. | Cook, T. R.; Stang, P. J. Chem. Rev. 2015, 115, 7001–7045. doi:10.1021/cr5005666 |
| 29. | Zhang, Y.-Y.; Gao, W.-X.; Lin, L.; Jin, G.-X. Coord. Chem. Rev. 2017, 344, 323–344. doi:10.1016/j.ccr.2016.09.010 |
| 30. | Wu, G.-Y.; Chen, L.-J.; Xu, L.; Zhao, X.-L.; Yang, H.-B. Coord. Chem. Rev. 2018, 369, 39–75. doi:10.1016/j.ccr.2018.05.009 |
| 40. | Liu, X.; Liu, H.; Zhou, W.; Zheng, H.; Yin, X.; Li, Y.; Guo, Y.; Zhu, M.; Ouyang, C.; Zhu, D.; Xia, A. Langmuir 2010, 26, 3179–3185. doi:10.1021/la903838w |
| 33. | Han, Y.-F.; Lin, Y.-J.; Jia, W.-G.; Jin, G.-X. Organometallics 2008, 27, 4088–4097. doi:10.1021/om800426e |
| 25. | Lang, J.-P.; Xu, Q.-F.; Yuan, R.-X.; Abrahams, B. F. Angew. Chem., Int. Ed. 2004, 43, 4741–4745. doi:10.1002/anie.200460076 |
| 33. | Han, Y.-F.; Lin, Y.-J.; Jia, W.-G.; Jin, G.-X. Organometallics 2008, 27, 4088–4097. doi:10.1021/om800426e |
| 6. | Cook, T. R.; Vajpayee, V.; Lee, M. H.; Stang, P. J.; Chi, K.-W. Acc. Chem. Res. 2013, 46, 2464–2474. doi:10.1021/ar400010v |
| 7. | Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j |
| 8. | Han, Y.-F.; Jin, G.-X. Chem. Soc. Rev. 2014, 43, 2799–2823. doi:10.1039/C3CS60343A |
| 9. | Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571–3579. doi:10.1021/ar500335a |
| 41. | Min, D.; Cho, B.-Y.; Lee, S. W. Inorg. Chim. Acta 2006, 359, 577–584. doi:10.1016/j.ica.2005.09.041 |
© 2018 Ma et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)