Search results
Search for "calculations" in Full Text gives 705 result(s) in Beilstein Journal of Nanotechnology. Showing first 200.
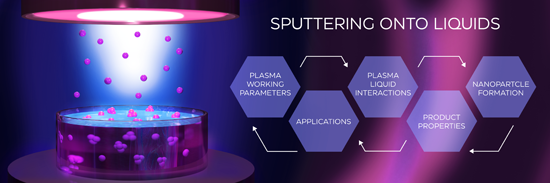
Sputtering onto liquids: a critical review
Beilstein J. Nanotechnol. 2022, 13, 10–53, doi:10.3762/bjnano.13.2






























Heating ability of elongated magnetic nanoparticles
Beilstein J. Nanotechnol. 2021, 12, 1404–1412, doi:10.3762/bjnano.12.104

- nanoparticles with aspect ratio a/b > 1. Although in some experimental works [31][33] it has been argued that an increase in the particle aspect ratio contributes to an increase in SAR, our calculations show that the change in the magnetic properties of an assembly as a function of the particle aspect ratio is
- ]: Here kB is the Boltzmann constant, γ is the gyromagnetic ratio, κ is phenomenological damping constant, δαβ is the Kronecker delta, and δ(t) is the delta function. The calculations of the low-frequency hysteresis loops are carried out at room temperature, T = 300 K. Results and Discussion SAR of dilute
- the viscosity of the medium where the nanoparticles are distributed. However, the orientation of the cubic easy anisotropy axes of magnetite nanoparticles can hardly be changed. In the calculations performed it is supposed to be random as before. In Figure 3a solid curves show the low-frequency





Identifying diverse metal oxide nanomaterials with lethal effects on embryonic zebrafish using machine learning
Beilstein J. Nanotechnol. 2021, 12, 1297–1325, doi:10.3762/bjnano.12.97

- surface functional groups [31]. (All calculations treated the core material and surface components independently, that is, the descriptors were calculated for the surface components treated as free molecules.) Since shell and/or surface functional groups were not present for all ENMs (i.e., the ENM could
- the sections “Descriptor calculations”, “Inapplicable descriptor dummy values” and “Descriptor pre-processing” under the Experimental section for full details.) First, the presence or absence of organic surface components was represented using binary indicator variables. Their negligible significance











Nonmonotonous temperature dependence of Shapiro steps in YBCO grain boundary junctions
Beilstein J. Nanotechnol. 2021, 12, 1279–1285, doi:10.3762/bjnano.12.95

- are in agreement with the calculations based on the resistively–capacitively shunted junction model and Bessel theory. The emergence of the receiving optima is explained by the mutual influence of the varying critical current and the characteristic frequency. Keywords: characteristic frequency
- discussed, and the measurement results are compared with the results of numerical calculations. Experimental Setup and Numerical Model The samples of grain boundary Josephson junctions were fabricated by on-axis dc magnetron sputtering [28][29][30][31] of YBa2Cu3O7−δ (YBCO) film on the surface of 24°[001
- function, which, in the first approximation, determines the response of the JJ to a change in the gigahertz-signal power. Equation 4 shows that as ωc grows, the Bessel function period increases, that is, the derivative dΔI1/dPmw decreases. Figure 5 shows the results of the numerical calculations of the





Impact of electron–phonon coupling on electron transport through T-shaped arrangements of quantum dots in the Kondo regime
Beilstein J. Nanotechnol. 2021, 12, 1209–1225, doi:10.3762/bjnano.12.89

- systems close to the unitary Kondo limit. However, due to its simplicity, this approach is also often used in analysis of the linear conductance of systems with weakly broken symmetry giving results in a reasonable agreement with experiment and with numerical renormalization group calculations [67]. Mean
- intermediate coupling is observed λ ≈ 0.5 [45], and for different C140 samples λ ranges between 0.1 and 4 [87][88]. In this section we present numerical calculations illustrating the effect of phonons on interference and electron correlations and we show how this effect is reflected in the conductance values
- ], that emergent low-energy SU(4) symmetry can be restored also for slightly asymmetric systems by appropriately adjusting the gate voltages. More recent analysis based on NRG calculations [91] showed however, that for U’ < U the restoration of symmetry in the low-energy range might only happen if the











Irradiation-driven molecular dynamics simulation of the FEBID process for Pt(PF3)4
Beilstein J. Nanotechnol. 2021, 12, 1151–1172, doi:10.3762/bjnano.12.86

- and dynamics of complex “meso-bio-nano” systems [14]. MBN Studio is a powerful multi-task toolkit enabling to set up and start MBN Explorer calculations, to monitor their progress, to examine calculation results, to visualize inputs and outputs, and to analyze specific characteristics determined by
- ://webbook.nist.gov/) and PubChem (https://pubchem.ncbi.nlm.nih.gov/), or can be determined via DFT calculations [11][12][29]. MBN Explorer and MBN Studio enable the creation of various crystalline substrates for which the unit cell and translation vectors are specified. The software tools enable also creating
- Pt(PF3)4 molecule is determined via DFT calculations and then optimized using MBN Explorer. The rCHARMM parameters for a Pt(PF3)4 molecule are determined from a series of DFT-based potential energy scans, similar to how it was done in [29] for a W(CO)6 precursor molecule. In brief, the DFT












Open-loop amplitude-modulation Kelvin probe force microscopy operated in single-pass PeakForce tapping mode
Beilstein J. Nanotechnol. 2021, 12, 1115–1126, doi:10.3762/bjnano.12.83

- and small-amplitude ringing was low-pass filtered at 25 kHz, which is a few times larger than the bias frequency, such that the CPD calculations will not be affected by this filter, but below the first resonance frequency to remove the induced ringing. The filtered signal (red curve in Figure 2








First-principles study of the structural, optoelectronic and thermophysical properties of the π-SnSe for thermoelectric applications
Beilstein J. Nanotechnol. 2021, 12, 1101–1114, doi:10.3762/bjnano.12.82

- (DFT). Our DFT calculations reveal that π-SnSe features an optical bandgap of 1.41 eV and has an exceptionally large lattice constant (12.2 Å, P213). We report several thermodynamic, optical, and thermoelectric properties of this π-SnSe phase for the first time. Our finding shows that the π-SnSe alloy
- ) calculations along with an experimental characterization they concluded that π-SnSe is energetically close to the stable α-SnSe phase. A working solar cell made by a cubic SnS crystalline structure is also reported [48] with optical bandgaps in the range of 1.6 to 1.8 eV [49]. Therefore, it is natural to
- thermophysical properties of the π-SnSe alloy, we have used first-principles calculations based on the full-potential linearized augmented plane-wave (FP-LAPW) method, implemented in the WIEN2k code [50][51]. A preliminary crystal lattice is initially made by using the details presented in [46] and then atomic












Criteria ruling particle agglomeration
Beilstein J. Nanotechnol. 2021, 12, 1093–1100, doi:10.3762/bjnano.12.81

- random process driven in the direction of a maximum of the entropy. Such a process can be simulated numerically using Monte Carlo calculations. The basic idea for such an algorithm consists of the following steps: As basic assumption, an ensemble of Ntot equal particles is selected. As these particles
- step, one reduces the number of elements, particles plus agglomerates, in the distribution by one. This process is repeated. The corresponding algorithm is sketched in Figure 1. To obtain statistically significant results, the calculations were performed with 105 and 107 particles in the starting
- increase of the entropy had to be removed. Obviously, because of the fluctuations, an algorithm as explained above does not allow for a determination of the maximum level of the entropy. As result of the calculations, one obtains a correlation between the number of collisions and the resulting particle







A new method for obtaining model-free viscoelastic material properties from atomic force microscopy experiments using discrete integral transform techniques
Beilstein J. Nanotechnol. 2021, 12, 1063–1077, doi:10.3762/bjnano.12.79

- calculations. Specifically, modern computational coding languages use variable precision. For example, for a number that is of order unity, the computational precision is ≈10−16, but for a number that is of order 10100 the precision is ≈1084. Therefore, in calculations with large numbers, one can accumulate a
- Fourier transforms can be found in Supporting Information File 1, Figure S9. (a, d) Amplitude of simulated retardance in the z-domain for an AFM simulation. (b, e) Retardance of the material calculated from the theoretical values. (c, f) Error between the simulation and the theoretical calculations. The











Revealing the formation mechanism and band gap tuning of Sb2S3 nanoparticles
Beilstein J. Nanotechnol. 2021, 12, 1021–1033, doi:10.3762/bjnano.12.76

- can mathematically be treated as a material with an indirect allowed transition. For crystalline Sb2S3, Filip et al. [47] and Vadapoo et al. [48] did first-principle calculations of the band structures. Both found indirect transitions as energetically most favorable but with only a little difference
- direct one in the present work. In contrast, Validžić et al. performed calculations based on the density functional theory and found a direct band gap [31]. The optical data can be seen in Figure 9 and in Figure S9 in Supporting Information File 1. As the spectra of the 2 h, 5 h, and 8 h samples are









Is the Ne operation of the helium ion microscope suitable for electron backscatter diffraction sample preparation?
Beilstein J. Nanotechnol. 2021, 12, 965–983, doi:10.3762/bjnano.12.73

- to the interaction volume depth. This is referred to as the steady-state condition. The time it takes to mill the sample to that depth and until the steady-state condition is reached determines the maximal area of specific ion implantation concentration. The calculations presented here determine the
- . The darker grains are the slower milling grains [11] which can accumulate a higher Ga impurity concentration according to the calculations above. To verify this, EDS measurements (see Supporting Information File 3) were performed on a Ga irradiated area on slower and faster milling grains. These
- the steady-state condition is reached later, allowing for a higher Ga impurity implantation concentration. The experimental result is in good agreement with the theoretical prediction from the Monte Carlo simulation and subsequent calculations. The Cu3Ga phase-transformed regions are larger for the









Prediction of Co and Ru nanocluster morphology on 2D MoS2 from interaction energies
Beilstein J. Nanotechnol. 2021, 12, 704–724, doi:10.3762/bjnano.12.56

- preferred. Predictions made using these descriptors can be used when deciding which metal–substrate combination will be suitable for a particular application where the shape of the metal is vital. Methods All calculations for this study were carried out with density functional theory (DFT) using the Vienna











Nanoporous and nonporous conjugated donor–acceptor polymer semiconductors for photocatalytic hydrogen production
Beilstein J. Nanotechnol. 2021, 12, 607–623, doi:10.3762/bjnano.12.50

- activity than double F substitution, and the polymer with the methoxy group at the meta site and the F atom at the para site yields the highest HER due to the excellent mobility of photogenerated charge carriers and the broad light absorption range. DFT calculations also verified that incorporating F on
- with pyrene and BT exhibited the best HER (177.50 μmol·h−1, 20 mg) due to low charge recombination and strong photoinduced charge transfer. Furthermore, DFT calculations (Figure 7) indicated that incorporating halogen atoms in both P43 and P44 (Figure 5) reduces the energy barrier for forming H
- hyperbranched Pdots. Araujo et al. [72] studied several BT-containing small molecules with A–D–A architectures through theoretical calculations. They found that small molecules could also mediate photocatalytic water splitting into hydrogen and oxygen. Dibenzothiophene-S,S-dioxide-based conjugated polymers










Stability and activity of platinum nanoparticles in the oxygen electroreduction reaction: is size or uniformity of primary importance?
Beilstein J. Nanotechnol. 2021, 12, 593–606, doi:10.3762/bjnano.12.49

- of the NP shape, leading to an increase in the proportion of more active areas, can lead to a significant increase in the Pt/C activity [18]. Moreover, according to the calculations in [17], NPs of each size can have its own optimal shape, which provides the highest ORR mass activity. Nevertheless









Properties of graphene deposited on GaN nanowires: influence of nanowire roughness, self-induced nanogating and defects
Beilstein J. Nanotechnol. 2021, 12, 566–577, doi:10.3762/bjnano.12.47

- graphene deposited on gallium nitride nanowires (GaN NWs) with different variations in height. The electric field induced in GaN predicted by theoretical calculations could reach 5 MV/cm [21]. This is an effect of high spontaneous and piezoelectric polarisations in the wurtzite structure of GaN
- the value of RDD’ ratio equals to 3.5 is characteristic of grain boundaries, five is characteristic of multiple vacancies, seven corresponds to single vacancies, while 13 is observed for sp3 hybridisation defects [20][49]. Furthermore, theoretical calculations predicted values of 1.3 and 10.5 for on








Surface-enhanced Raman scattering of water in aqueous dispersions of silver nanoparticles
Beilstein J. Nanotechnol. 2021, 12, 497–506, doi:10.3762/bjnano.12.40

- was found that only one water molecule per 10 million is in direct contact with the surface of AgNPs. It is also worthy to mention that the model used to calculate the part of the water molecules in the direct vicinity of AgNPs yielded an overestimated value (the detailed calculations are described in
- calculate the EF. As all measurements were performed under the same conditions, all calculations, for simplicity, were done for 1 dm3 of the samples (water and AgNPs blue dispersion). The enhancement factor for AgNPs blue was calculated by using Equation 1 as follows: ISERS and IRS are integral intensities
- for AgNPs blue and water spectra, respectively (AgNPs blue spectrum after the reabsorption correction) and Nsurf and Nvol are the numbers of water molecules located on the surface of AgNPs and water molecules in pure water in 1 dm3, respectively (for the calculations of these values, see section SI3





Reconstruction of a 2D layer of KBr on Ir(111) and electromechanical alteration by graphene
Beilstein J. Nanotechnol. 2021, 12, 432–439, doi:10.3762/bjnano.12.35

- pattern. Comprehensive calculations by DFT, taking into account the observed periodicities, resulted in a new low-energy reconstruction. However, it is fully relaxed into a common cubic structure when a monolayer of graphene is located between substrate and KBr. By using Kelvin probe force microscopy, the
- work functions of the reconstructed and the cubic configuration of KBr were measured and indicate, in accordance with the DFT calculations, a difference of nearly 900 meV. The difference is due to the strong interaction and local charge displacement of the K+/Br− ions and the Ir(111) surface, which are
- temperature and density functional theory (DFT) calculations. The results suggest that this particular reconstruction of KBr occurs on Ir(111), due to a specific correlation of the lattice parameter. When deposited on a single layer of graphene on the same substrate, the topography of the KBr islands returns




Characterization, bio-uptake and toxicity of polymer-coated silver nanoparticles and their interaction with human peripheral blood mononuclear cells
Beilstein J. Nanotechnol. 2021, 12, 282–294, doi:10.3762/bjnano.12.23

- binding to proteins present in the RPMI. Formation of dissolved silver chloride was suggested by speciation calculations (data not reported, as the accuracy of calculations can be questioned, given the media complexity and difference from those on which the model was validated). There is limited evidence
- sample of Person 5 showed a different trend in the LDH assay than the other samples and it is not clear if this discrepancy was a result of variability between individuals. Excluding the sample of Person 5 from the calculations, the Pearson correlation coefficient indicated a moderate correlation between






Determination of elastic moduli of elastic–plastic microspherical materials using nanoindentation simulation without mechanical polishing
Beilstein J. Nanotechnol. 2021, 12, 213–221, doi:10.3762/bjnano.12.17

- elastic modulus extracted according to Equation 8. Oliver and Pharr proposed that β = 1.05 with a potential error of approximately ±0.05, based on their analysis of available results 1.0226 ≤ β ≤ 1.085 from experiments and finite element calculations [6]. Finite Element Method To evaluate the mechanical







TiOx/Pt3Ti(111) surface-directed formation of electronically responsive supramolecular assemblies of tungsten oxide clusters
Beilstein J. Nanotechnol. 2021, 12, 203–212, doi:10.3762/bjnano.12.16

- supramolecular structures. It is noteworthy that individual W3O9 clusters, according to previous density functional theory (DFT) calculations [11], are characterized by the most stable six-membered ring structure with D3h symmetry. It consists of oxygen-bridged tungsten atoms with two additional terminal oxygen
- with a constant distance between them. The DFT calculations of a very similar oxide phase, grown by a reactive Ti deposition on Pt(111) [25], showed that the brightest spots belong to fourfold oxygen-coordinated titanium atoms, whereas threefold coordinated Ti atoms are depicted with less contrast. The
- atoms remain in the oxidation state +VI and one is reduced to +IV (compare Figure 3c and Figure 3d). The latter one has less unoccupied electronic states and, therefore, is not resolved by scanning with a positive bias voltage. Our DFT calculations of W3O9 and of a hypothetical W3O8 molecule in the gas





Mapping the local dielectric constant of a biological nanostructured system
Beilstein J. Nanotechnol. 2021, 12, 139–150, doi:10.3762/bjnano.12.11

- FIB. Capacitance model with tip, sample, conductive plate, and the parameters used in our calculations. R is the tip apex radius, θ is the tip conical angle, z is the tip–sample distance, h is the sample thickness and εr is the relative permittivity of the sample. The sample is shown on top of the









Numerical analysis of vibration modes of a qPlus sensor with a long tip
Beilstein J. Nanotechnol. 2021, 12, 82–92, doi:10.3762/bjnano.12.7

- of the tuning fork prong without a tip. Figure 2b visualizes the mesh distribution used for the calculations. The mesh density increases at the link between the two prongs and near the boundaries of each material. Figure 2c is a zoomed view of the tungsten wire attached to the end of the tuning fork
- case, the volume of the glue in the simulation is approximately equal to the volume of the glue in the experiment. Figure 2d is a false-color representation of the total displacement levels. Warmer colors represent a larger displacement. All calculations were carried out using COMSOL Multiphysics
- used for finite element calculations. Supporting Information Supporting Information File 32: Additional simulation results. Funding The authors gratefully acknowledge the support from the National Key Technologies R&D Program of China (Grant No. 2016YFA0201101), the National Natural Science









Free and partially encapsulated manganese ferrite nanoparticles in multiwall carbon nanotubes
Beilstein J. Nanotechnol. 2020, 11, 1891–1904, doi:10.3762/bjnano.11.170

- , experimentally, bulk MnFe2O4 is known to have semiconductor properties [18]. The spectrum shown in Figure 1e contains a main peak at approx. 5.4 eV and weaker peaks at approx. 9.7 eV and 12.1 eV. By comparing the spectrum with the band structure calculations, the first peak (indicated by an arrow in the spectrum









Kondo effects in small-bandgap carbon nanotube quantum dots
Beilstein J. Nanotechnol. 2020, 11, 1873–1890, doi:10.3762/bjnano.11.169

- changes of the bandgap. In the following we show how, for a given nearly metallic nanotube, one can change the position of high-symmetry points by strain and a magnetic field. Our calculations also show that in a quantum dot formed in a small-bandgap nanotube electron and hole states can degenerate in
- Fermi velocity (vF ≅ 0.8c), D is the nanotube diameter, and a is the distance between carbon atoms in the A lattice and the B lattice of graphene (a ≅ 0.254 nm). Eg is the bandgap energy, where Θ is the chiral angle, . According to tight-binding calculations the value of β corresponding to the
- calculations [27][28]. The SBMFA method best describes systems close to the Kondo fixed point, that is, for the case of deep fully degenerate atomic levels at low temperatures. This approximation correctly describes spin or pseudospin fluctuations and reproduces local Fermi-liquid behavior at zero temperature






![[Graphic 37]](/bjnano/content/inline/2190-4286-11-169-i52.svg?max-width=637&scale=1.18182) as a functio...
as a functio...




