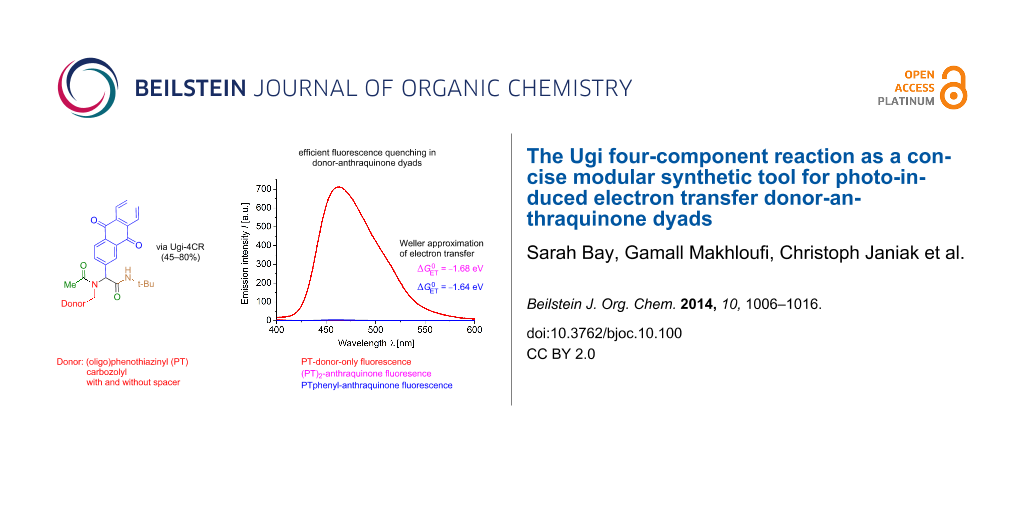
Abstract
Phenothiazinyl and carbazolyl-donor moieties can be covalently coupled to an anthraquinone acceptor unit through an Ugi four-component reaction in a rapid, highly convergent fashion and with moderate to good yields. These novel donor–acceptor dyads are electronically decoupled in the electronic ground state according to UV–vis spectroscopy and cyclic voltammetry. However, in the excited state the inherent donor luminescence is efficiently quenched. Previously performed femtosecond spectroscopic measurements account for a rapid exergonic depopulation of the excited singlet states into a charge-separated state. Calculations of the Gibbs energy of photo-induced electron transfer from readily available UV–vis spectroscopic and cyclovoltammetric data applying the Weller approximation enables a quick evaluation of these novel donor–acceptor dyads. In addition, the X-ray structure of a phenothiazinyl–anthraquinone dyad supports short donor–acceptor distances by an intramolecular π-stacking conformation, an important assumption also implied in the calculations of the Gibbs energies according to the Weller approximation.

Graphical Abstract
Introduction
Chromophores, fluorophores, and electrophores, are functional organic materials [1] and constitute active components in molecular electronics [2], photonics [3], and bioanalytics [4-6]. Therefore, the design of well-defined monomolecular structures with electron-donor (Do) and acceptor (Acc) substitution, so called Do–Acc dyads, is a topical field with a paramount academic and technological interest [7,8]. Tailor-made Do–Acc systems represent the fundamental basis for application in molecular electronics and optoelectronics [9-14] and they are employed in organic light-emitting diodes (OLEDs) for a balanced charge transport [15-20] and photovoltaic devices [21-25]. The concept of persistent light-induced charge separation between a donor and an acceptor originates from photosynthesis, nature’s most important process to convert sunlight into chemical energy. One of the most challenging endeavors of mankind is the unlimited generation of electrical energy from sunlight, with great efforts to mimicking photosynthesis by creation of artificial photosynthetic systems [26,27]. The simulation of relevant processes has reached a high level of understanding and the primary process of light-induced charge separation in various types of Do–Acc dyads has been intensively studied [28,29]. This photo-induced electron transfer (PET) [30-34] has been investigated with donors such as porphyrines, polycyclic aromatic hydrocarbons, perylenediimides and (oligo)thiophenes [35,36], tetrathiafulvalenes [37], as well as phenothiazine and its derivatives [22,38-40]. The latter have become attractive electrophores due to their reversible and tunable oxidation potential. Interestingly quenching of the phenothiazine inherent fluorescence offers a facile evidence for the occurrence of intramolecular PET in phenothiazine-containing Do–Acc dyads [41,42]. As suitable acceptor moieties C60 fullerene [43-45], and quinones, such as 9,10-anthraquinone as a potential two electron acceptor, have been commonly used in Do–Acc arrangements [46-51]. In previous studies phenothiazine–anthraquinone couples have been introduced into peptide scaffolds [52-54] and rigid Do–Acc dyads [55]. Nevertheless, a modular and rapid access by multicomponent reactions to these types of functional targets has never been explored prior to our recent studies [56]. For instance, the Ugi four-component reaction (Ugi 4CR) [57-60] establishes the chemically robust α-aminoacylamide backbone in one step and with high diversity. Therefore, it is also extensively used in medicinal and combinatorial chemistry for lead finding and optimization [61]. We have recently employed the Ugi 4CR as a one-step process to simultaneously introduce a phenothiazine-functionalized amine and an anthraquinone-substituted aldehyde together with acetic acid and tert-butyl isocyanide for rapidly assembling a donor–acceptor conjugate 1 displaying a photo-induced electron transfer leading to a charge-separated state with a lifetime of >2 ns (Figure 1), as elucidated by femtosecond transient absorption spectroscopy [56]. The donor-only (2) and acceptor-only (3) models for spectroscopic comparison were obtained analogously.
![[1860-5397-10-100-1]](/bjoc/content/figures/1860-5397-10-100-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Phenothiazine–anthraquinone dyad 1, donor-only (2) and acceptor-only (3) models assembled by Ugi 4CR.
Figure 1: Phenothiazine–anthraquinone dyad 1, donor-only (2) and acceptor-only (3) models assembled by Ugi 4C...
For enabling rapid accesses to functional π-systems, such as Do–Acc dyads for photo-induced charge separation, besides a robust, flexible diversity-oriented one-pot reaction, such as the Ugi 4CR, a quick semiquantitative estimation of the feasibility of the charge-separated state based upon inexpensive analytical methods is highly desirable. Here we present the synthetic versatility of this multicomponent approach to Do–anthraquinone dyads, as exemplified by phenothiazinyl and carbazole moieties as donors, and comprehensive physical organic studies of electronic and electrochemical properties investigated by steady state UV–vis and fluorescence spectroscopy as well as cyclic voltammetry. The obtained data are interpreted in the light of the Weller approximation to estimate the probability for charge separation by photo-induced electron transfer based upon its Gibbs energy calculated from the analytical data and donor–acceptor distances of lowest energy conformers from inexpensive force field computations.
Results and Discussion
Synthesis and structure
Within the concept of diversity-oriented syntheses of chromophores [62-69] we have established accesses to chromophores in a one-pot fashion based upon transition metal catalysis as an entry to consecutive multicomponent [70,71] and domino reactions [72]. The highly convergent synthetic approach by multicomponent reactions should as well be applicable to functional Do–Acc dyads. Therefore, we set out to place electron-rich phenothiazinyl and carbazolyl derivatives 4 as amino component in the Ugi 4CR, whereas the electron acceptor was introduced as anthraquinone-2-carbaldehyde (5). Acetic acid (6) and tert-butyl isocyanide (7) were the two residual components (Scheme 1) [56].
![[1860-5397-10-100-i1]](/bjoc/content/inline/1860-5397-10-100-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Ugi 4CR synthesis of donor–anthraquinone dyads 8.
Scheme 1: Ugi 4CR synthesis of donor–anthraquinone dyads 8.
The most favorable solvent for Ugi 4CR is methanol. However, to increase solubility portions of dichloromethane were added to assure a homogeneous solution. For liberating the free base from methylamine hydrochlorides 4, potassium hydroxide was employed as a base. The methylamine hydrochlorides 4, in turn, were readily available from the corresponding cyano compounds [73] by lithium alanate reduction in diethyl ether [74]. The corresponding donor–anthraquinone dyads 8 were isolated in moderate to good yields. For reference, donor-only systems 10 were also prepared by Ugi 4CR from the amino derivatives 4 and acetaldehyde (9), in moderate to good yield, with acetic acid (6) and tert-butyl isocyanide (7) as the corresponding acid and isonitrile components (Scheme 2).
![[1860-5397-10-100-i2]](/bjoc/content/inline/1860-5397-10-100-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Ugi 4CR synthesis of donor-only reference systems 10.
Scheme 2: Ugi 4CR synthesis of donor-only reference systems 10.
The appearance of single signal sets in the 1H and 13C NMR spectra of 8 and 10 unambiguously supports the structural assignment and that isomeric mixtures due to restricted amide-bond rotation can be excluded. Besides mass spectrometry and combustion analysis the structure of phenothiazine–anthraquinone dyads 8a–d was additionally supported by an X-ray structure analysis of the partially oxidized derivative of compound S(O)-1 (Figure 2) [75]. The phenothiazine and anthraquinone moieties are aligned by intramolecular π-stacking with an average distance of ~3.9 Å [76,77]. In the unit cell the R- and S-enantiomers of a single diastereomer (S-oxide) are arranged in pairs resulting in four stacked (hetero)aromatic units (Figure 2), so that the anthraquinone moieties of two molecules display an average distance of ~3.8 Å. Based upon the X-ray data quantum mechanical computations on this conformer were envisioned (vide infra).
![[1860-5397-10-100-2]](/bjoc/content/figures/1860-5397-10-100-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of S(O)-1 (left) (30% ellipsoids, except for the CH3CH2 end of the hexyl group, the disordered water molecules were omitted for clarity) and the intra- and intermolecular π–π-stacking interactions between an inversion symmetry related pair (right, hydrogen atoms were omitted for clarity).
Figure 2: Molecular structure of S(O)-1 (left) (30% ellipsoids, except for the CH3CH2 end of the hexyl group,...
Electronic properties and electronic structure
All dyads 8 and 1 as well as the reference systems 2, 3, and 10 were studied by cyclic voltammetry in dichloromethane at room temperature (Table 1, Figure 3).
Table 1: Cyclovoltammetric data of the dyads 8 and the reference systems 1, 2, 3 and 10 (recorded in CH2Cl2, T = 298 K, c = 0.1 mol·L−1, Pt working electrode, Pt counter electrode, Ag/AgCl reference electrode, electrolyte N(n-Bu)4PF6, scan rates v of 100, 250, 500 and 1000 mV·s−1)a,b.
| Compound | E1/20/+1 [mV] | E1/2+1/+2 [mV] | E1/2−1/0 [mV] | E1/2−2/−1 [mV] |
|---|---|---|---|---|
| 8a | 780 | – | −850c | −1500c |
| 8b | 780 | – | −870c | −1370c |
| 8c | 710 | 1450 | −900c | −1390c |
| 8d | 630 | 800 | −880c | −1380c |
| 8e | 1220c | – | −940c | −1420c |
| 8f | 1170c | – | −920c | – |
| 1 | 710 | – | −870c | −1430c |
| 3 | – | – | −920c | −1430c |
| 2 | 730 | – | – | – |
| 10a | 710 | 1430 | – | – |
| 10b | 1320c | – | – | – |
aCalibrated against ferrocene as an internal standard (E00/+1 = 450 mV). bThe half-wave potentials E1/2 were extrapolated to a scan rate of v = 0 mV·s−1 from the linear correlation plot of the differences of the anodic and cathodic peak potentials against ![[Graphic 13]](/bjoc/content/inline/1860-5397-10-100-i17.svg?max-width=637&scale=1.18182) . cQuasi-reversible redox wave.
. cQuasi-reversible redox wave.
![[1860-5397-10-100-3]](/bjoc/content/figures/1860-5397-10-100-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Cyclic voltammogram of dyad 8c (recorded in CH2Cl2, T = 298 K, c (8c) = 0.1 mol·L−1, Pt working electrode, Pt counter electrode, Ag/AgCl reference electrode, electrolyte N(n-Bu)4PF6, scan rate of 250 mV·s−1).
Figure 3: Cyclic voltammogram of dyad 8c (recorded in CH2Cl2, T = 298 K, c (8c) = 0.1 mol·L−1, Pt working ele...
The cyclic voltammograms were recorded at scan rates v of 100, 250, 500 and 1000 mV·s−1 and the differences of anodic and cathodic peak potentials were plotted against ![[Graphic 12]](/bjoc/content/inline/1860-5397-10-100-i16.svg?max-width=637&scale=1.18182) for extrapolating the half-wave potentials E1/2 for a scan rate v = 0 mV·s−1 assuming an ideal Nernstian behavior. In the cyclic voltammograms of the phenothiazine–anthraquinone dyads 8a–d, typical for phenothiazine derivatives [41,42,78], first reversible oxidations E1/20/+1 between 630 and 780 mV are found, and in addition the cyclic voltammograms of 8c and 8d display second oxidation waves E1/2+1/+2 at 1450 (8c) and 800 mV (8d). The direct comparison of the 3-phenyl derivative 8c with the donor-only reference 10a clearly indicates that the proximity of the electron-withdrawing anthraquinone moiety does not affect the first and second reversible oxidations of the phenothiazinyl moiety in the Do–Acc dyad 8c. The system 8d containing two conjugated phenothiazinyl moieties is particular since the second oxidation wave originates from the electronic coupling within the diphenothiazine unit [79,80]. These values for first and second oxidation can also be found in the donor-only reference compound 10a. Furthermore two quasi-reversible reductions stemming from the anthraquinone core can be detected for E1/2−1/0 between −850 to −900 mV and for E1/2−2/−1 between −1370 and −1500 mV, in good agreement with literature data [81,82]. Within a margin of 70 mV both reduction waves fall into the same region as for the anthraquinone-only reference 3 with E1/2−1/0 at −920 mV and E1/2−2/−1 at −1430 mV. Cum grano salis the electrochemical behavior of the novel dyads 8a–d are very similar to the parent system 1. The carbazole-based dyads 8e and 8f only show quasi-reversible oxidation waves at an estimated E1/20/+1 of 1220 and 1170 mV, yet in good agreement with the behavior of the carbazole-only reference 10b with an estimated E1/20/+1 at 1320 mV. The carbazole–anthraquinone dyads 8e and 8f display the anthraquinone-centered first quasi-reversible reduction wave E1/2−1/0 at −940 (8e) and −920 mV (8f), the second quasi-reversible reduction wave is only found for dyad 8e and appears at E1/2−2/−1 = −1420 mV. The comparison of the cyclic voltammograms between all dyads 8 and the reference systems 2, 3, and 10 clearly shows that the donor and anthraquinone moieties are essentially electronically decoupled in the electronic ground state. Therefore, in the electronic ground state the electronic effects should behave additively, i.e., as if the donors and anthraquinones were placed at large distances.
for extrapolating the half-wave potentials E1/2 for a scan rate v = 0 mV·s−1 assuming an ideal Nernstian behavior. In the cyclic voltammograms of the phenothiazine–anthraquinone dyads 8a–d, typical for phenothiazine derivatives [41,42,78], first reversible oxidations E1/20/+1 between 630 and 780 mV are found, and in addition the cyclic voltammograms of 8c and 8d display second oxidation waves E1/2+1/+2 at 1450 (8c) and 800 mV (8d). The direct comparison of the 3-phenyl derivative 8c with the donor-only reference 10a clearly indicates that the proximity of the electron-withdrawing anthraquinone moiety does not affect the first and second reversible oxidations of the phenothiazinyl moiety in the Do–Acc dyad 8c. The system 8d containing two conjugated phenothiazinyl moieties is particular since the second oxidation wave originates from the electronic coupling within the diphenothiazine unit [79,80]. These values for first and second oxidation can also be found in the donor-only reference compound 10a. Furthermore two quasi-reversible reductions stemming from the anthraquinone core can be detected for E1/2−1/0 between −850 to −900 mV and for E1/2−2/−1 between −1370 and −1500 mV, in good agreement with literature data [81,82]. Within a margin of 70 mV both reduction waves fall into the same region as for the anthraquinone-only reference 3 with E1/2−1/0 at −920 mV and E1/2−2/−1 at −1430 mV. Cum grano salis the electrochemical behavior of the novel dyads 8a–d are very similar to the parent system 1. The carbazole-based dyads 8e and 8f only show quasi-reversible oxidation waves at an estimated E1/20/+1 of 1220 and 1170 mV, yet in good agreement with the behavior of the carbazole-only reference 10b with an estimated E1/20/+1 at 1320 mV. The carbazole–anthraquinone dyads 8e and 8f display the anthraquinone-centered first quasi-reversible reduction wave E1/2−1/0 at −940 (8e) and −920 mV (8f), the second quasi-reversible reduction wave is only found for dyad 8e and appears at E1/2−2/−1 = −1420 mV. The comparison of the cyclic voltammograms between all dyads 8 and the reference systems 2, 3, and 10 clearly shows that the donor and anthraquinone moieties are essentially electronically decoupled in the electronic ground state. Therefore, in the electronic ground state the electronic effects should behave additively, i.e., as if the donors and anthraquinones were placed at large distances.
Based on the starting geometry from the X-ray structure analysis of S(O)-1 the frontier molecular orbitals (FMO) of 1 were calculated on the DFT level of theory with the B3LYP functional and the Pople basis set 6-311G* (Figure 4) [83-86]. It is noticeable that the coefficient density of the HOMO is almost completely localized on the phenothiazine unit whereas the coefficient density of the LUMO resides on the anthraquinone core, supporting the electronic decoupling of the donor and the acceptor in the electronic ground state. In conclusion the computation underlines that in dyad 1 an electronic prerequisite for electronically favored electron-transfer processes in donor–acceptor systems is the spatial proximity of PT and AQ. This conformer is additionally stabilized by π-stacking of the donor and the acceptor, which is adopted in the solid state and results in a strong bathochromically shifted absorption of the solid.
![[1860-5397-10-100-4]](/bjoc/content/figures/1860-5397-10-100-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: DFT-computed (B3LYP, 6-311G*) frontier molecular orbitals HOMO (bottom) and LUMO (top) of the phenothiazine–anthraquinone dyad 1.
Figure 4: DFT-computed (B3LYP, 6-311G*) frontier molecular orbitals HOMO (bottom) and LUMO (top) of the pheno...
Furthermore, the electronic properties of the donor-anthraquinone dyads 8 were studied by absorption and emission spectroscopy (Table 2). All phenothiazine-based dyads 8a–d show very similar absorption characteristics with a major absorption band around 259 nm and a lower intensity band around 325 nm (Figure 5). According to the phenothiazine-only (10a) and anthraquinone-only (3) references phenothiazine as well as anthraquinone absorb in the same region. In the spectrum of the carbazole dyad 8e the carbazole-typical absorption maxima can be found (cf reference systems 3 and 10b), whereas the spectrum of 8f displays just two absorption bands at 255 and 285 nm, originating from the 3-phenylcarbazole moiety.
Table 2: Absorption and emission characteristics of the dyads 8, 1, and the reference systems 2, 3, and 10 (recorded in CH2Cl2, c = 1.4–3.1∙10−5 mol·L−1, T = 298 K).
| Compound | Absorption | Emission | Stokes shift |
|---|---|---|---|
| λmax,abs [nm] (ε [L·mol·cm−1]) | λmax,em [nm] |
Δ![[Graphic 1]](/bjoc/content/inline/1860-5397-10-100-i5.svg?max-width=637&scale=1.18182) [cm−1]a [cm−1]a
|
|
| 8a | 257 (44000), 319 (5000) | –b | – |
| 8b | 258 (87000), 325 (10000) | –b | – |
| 8c | 258 (92000), 326 (19000) | –b | – |
| 8d | 259 (145000), 326 (40000) | –b | – |
| 8e | 250 (58000), 265 (69000), 298 (21000), 335 (11000) | –b | – |
| 8f | 255 (85000), 285 (62000) | –b | – |
| 1 | 258 (69000), 323 (11000) | –b | – |
| 3 | 259 (49000), 329 (6000) | –b | – |
| 2 | 259 (39000), 311 (6000) | 450c | 9900 |
| 10a | 269 (61000), 320 (18000) | 463d | 9700 |
| 10b | 240 (47000), 267 (29000), 298 (17000), 336 (4000), 352 (4000) | 362, 377e | 800 |
aΔ![[Graphic 2]](/bjoc/content/inline/1860-5397-10-100-i6.svg?max-width=637&scale=1.18182) = 1/λmax,abs − 1/λmax,em [cm−1]. bThe residual fluorescence is only detectable in the base line, i.e., less than 5% in a.u., vide infra. cλexc = 311 nm. dλexc = 320 nm. eλexc = 298 nm.
= 1/λmax,abs − 1/λmax,em [cm−1]. bThe residual fluorescence is only detectable in the base line, i.e., less than 5% in a.u., vide infra. cλexc = 311 nm. dλexc = 320 nm. eλexc = 298 nm.
![[1860-5397-10-100-5]](/bjoc/content/figures/1860-5397-10-100-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Normalized absorption spectra of the phenothiazine–anthraquinone dyad 8c (recorded in CH2Cl2, c (8c) = 2.5∙10−5 mol·L−1, T = 298 K).
Figure 5: Normalized absorption spectra of the phenothiazine–anthraquinone dyad 8c (recorded in CH2Cl2, c (8c...
The extinction coefficients of the Do–anthraquinone dyads 8 are expectedly larger than those of the donor-only (10) or anthraquinone-only (3) compounds. Plotting the extinction coefficient against the wavelength it becomes evident that the Do–anthraquinone dyads behave additively with respect to the constituent reference chromophores (Figure 6). Absorption spectroscopy as a probe for the electronic ground state also supports that in Do–anthraquinone dyads 8 the donor and anthraquinone moieties are electronically decoupled in the electronic ground state.
![[1860-5397-10-100-6]](/bjoc/content/figures/1860-5397-10-100-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Absorption spectra of Do–anthraquinone dyads 8c (top) and 8e (bottom) with the corresponding references 3 and 10, and their addition spectra (Do + Acc) (recorded in CH2Cl2, T = 298 K).
Figure 6: Absorption spectra of Do–anthraquinone dyads 8c (top) and 8e (bottom) with the corresponding refere...
Fluorescence is an excited-state phenomenon and, therefore, steady-state emission spectra of the donor-only reference systems 2 (Figure 7) and 10 and the donor–anthraquinone dyads 8 were recorded (Figure 8). For the donor–anthraquinone dyads 8 the emission is significantly quenched in comparison to the corresponding donor-only model systems 2 and 10 at the same concentrations according to relative fluorescence quantum yields Φf,rel (Table 3) and only residual weak emission traces from the donor and/or the anthraquinone excitations can be detected (for spectra of the residual emissions of the dyads 8 see Supporting Information File 1). Since the relative fluorescence quantum yields Φf,rel are attenuated by 95–99% in comparison to the donors’ fluorescence an efficient and rapid nonradiative depopulation of the excited state can be assumed by an electron transfer. This rationale is additionally supported by our previous transient absorption spectroscopic study of the dyad 1 [56].
![[1860-5397-10-100-7]](/bjoc/content/figures/1860-5397-10-100-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Normalized absorption and emission spectra of Ugi-donor compounds 2 and 10 (recorded in CH2Cl2, T = 298 K, λmax,exc (2) = 311 nm, λmax,exc (10a) = 320 nm, λmax,exc (10b) = 298 nm).
Figure 7: Normalized absorption and emission spectra of Ugi-donor compounds 2 and 10 (recorded in CH2Cl2, T =...
![[1860-5397-10-100-8]](/bjoc/content/figures/1860-5397-10-100-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Emission spectra of donor-only system 2 phenothiazine–anthraquinone dyads 8a,b (top), and the donor-only system 10a and the phenothiazine–anthraquinone dyads 8c,d (bottom) (recorded in CH2Cl2, T = 298 K, c = 0.7–2.9∙10−6 mol·L−1).
Figure 8: Emission spectra of donor-only system 2 phenothiazine–anthraquinone dyads 8a,b (top), and the donor...
Table 3: Relative quantum yields Φf,rel of Do-anthraquinone dyads 8 in comparison to their donor-only reference systems 2 and 10.
| Donor-only reference | 2 | 10a | 10b | |||
|---|---|---|---|---|---|---|
| Donor-anthraquinone dyad | 8a | 8b | 8c | 8d | 8e | 8f |
| Φf,rela | 0.04 | 0.05 | <0.01 | <0.01 | 0.01 | 0.04 |
aDetermined in CH2Cl2, T = 298 K, the quantum yield of the corresponding reference was set to 1.0.
According to the Weller approximation [87] the driving force for a photo-induced electron transfer leading to a charge-separated state that is responsible for the observed fluorescence quenching can be calculated from the measured electrochemical and spectroscopic data. Among several representations for calculating the Gibbs free energy of the electron transfer ![[Graphic 3]](/bjoc/content/inline/1860-5397-10-100-i7.svg?max-width=637&scale=1.18182) [88,89] can be described by Equation 1 [90]
[88,89] can be described by Equation 1 [90]
![[1860-5397-10-100-i3]](/bjoc/content/inline/1860-5397-10-100-i3.svg?max-width=590&scale=1.18182)
Where ![[Graphic 4]](/bjoc/content/inline/1860-5397-10-100-i8.svg?max-width=637&scale=1.18182) is the difference of the first oxidation potential of the donor and first reduction potential of the acceptor, respectively, E00 expresses the energy of the photonic excitation, RDo–Acc delineates the distance between the centers of the donor and acceptor moieties, εs and εref are the dielectric constants of the solvent applied in spectroscopy (εs) and reference solvent used in electrochemistry (εref), and r+ and r− are indicating the effective ionic radii of the donor radical cation and acceptor radical anion, respectively. It is allowed to neglect the forth term, if spectroscopic and electrochemical measurements are performed in the same solvent. Therefore, Equation 1 simplifies to Equation 2 for calculating
is the difference of the first oxidation potential of the donor and first reduction potential of the acceptor, respectively, E00 expresses the energy of the photonic excitation, RDo–Acc delineates the distance between the centers of the donor and acceptor moieties, εs and εref are the dielectric constants of the solvent applied in spectroscopy (εs) and reference solvent used in electrochemistry (εref), and r+ and r− are indicating the effective ionic radii of the donor radical cation and acceptor radical anion, respectively. It is allowed to neglect the forth term, if spectroscopic and electrochemical measurements are performed in the same solvent. Therefore, Equation 1 simplifies to Equation 2 for calculating ![[Graphic 5]](/bjoc/content/inline/1860-5397-10-100-i9.svg?max-width=637&scale=1.18182)
![[1860-5397-10-100-i4]](/bjoc/content/inline/1860-5397-10-100-i4.svg?max-width=590&scale=1.18182)
Where the first two terms indicate the free energy of the charge-separated state calculated from the spectroscopic and electrochemical measurements (in eV) and ![[Graphic 6]](/bjoc/content/inline/1860-5397-10-100-i10.svg?max-width=637&scale=1.18182) represents the correction term of the solvent polarity and the effect of the distance of the donor and acceptor moieties according to
represents the correction term of the solvent polarity and the effect of the distance of the donor and acceptor moieties according to ![[Graphic 7]](/bjoc/content/inline/1860-5397-10-100-i11.svg?max-width=637&scale=1.18182)
Indeed, for all Do-anthraquinone dyads 8 exergonic Gibbs free energies for the electron transfer are found, both for the simplified free enthalpy ΔGET,simpl, i.e., the first two terms of Equation 2, and upon taking solvation into account with the term ![[Graphic 8]](/bjoc/content/inline/1860-5397-10-100-i12.svg?max-width=637&scale=1.18182) . Therefore, the extent of the thermodynamically favored charge separation by an intramolecular photo-induced electron transfer (PET), plausibly explaining the observed fluorescence quenching, can be easily determined and compared within the series of related dyad systems (Table 4).
. Therefore, the extent of the thermodynamically favored charge separation by an intramolecular photo-induced electron transfer (PET), plausibly explaining the observed fluorescence quenching, can be easily determined and compared within the series of related dyad systems (Table 4).
Table 4:
Calculation of the Gibbs free energies for the simple electron transfer ΔGET,simpl, for the solvation![[Graphic 9]](/bjoc/content/inline/1860-5397-10-100-i13.svg?max-width=637&scale=1.18182) , and the electron transfer
, and the electron transfer ![[Graphic 10]](/bjoc/content/inline/1860-5397-10-100-i14.svg?max-width=637&scale=1.18182) according to the Weller approximation of the dyads 8.
according to the Weller approximation of the dyads 8.
| Compound | e[E0ox (Do) − E0red (Acc)] | E00 | ∆GET,simpl | RDo−Acc | ΔG0solv | ΔG0ET |
|---|---|---|---|---|---|---|
| [eV]a | [eV]b | [eV]c | [nm]d | [eV] | [eV] | |
| 8a | 1.60 | 3.08 | −1.46 | 0.39 | 0.40 | −1.78 |
| 8b | 1.65 | 3.08 | −1.43 | 0.43 | 0.37 | −1.66 |
| 8c | 1.51 | 2.93 | −1.42 | 0.53 | 0.30 | −1.68 |
| 8d | 1.61 | 2.93 | −1.32 | 0.38 | 0.41 | −1.64 |
| 8e | 2.16 | 3.49 | −1.34 | 0.40 | 0.39 | −1.70 |
| 8f | 2.09 | 3.49 | −1.41 | 0.46 | 0.34 | −1.69 |
aCalculated from (E1/20/+1 − E1/2−1/0) [V] obtained from cyclic voltammetry (see Table 1). bE00 [eV] of donor-only reference compounds 2 (E00 = 402 nm), 10a (E00 = 423 nm), and 10b (E00 = 355 nm) were estimated by the intersection of normalized absorption and emission spectra. c∆GET,simpl = e[E0ox (Do) − E0red (Acc)] − E00. dThe distances RDo−Acc [nm] of the donor and acceptor centers were estimated from lowest energy conformers by optimized MM2FF calculations [91] taking the distances between the centroid of the anthraquinone and the nitrogen atom of the donor moiety. For the diphenothiazine derivative 8d the more electron-rich inner phenothiazine was assumed to be oxidized first.
The negative free enthalpies of PET are numerically very similar for 8b, 8c, 8e, and 8f, however, larger in quantity for dyad 8a and smaller for dyad 8d. A diminishing of the distance between donor and anthraquinone, e.g., by adopting flexible close-contact conformations as for dyad 8a causes an increase in the driving force of the PET. Smaller excitation energies E00 and lower oxidation potentials as for the diphenothiazine dyad 8d cause a smaller PET driving force ![[Graphic 11]](/bjoc/content/inline/1860-5397-10-100-i15.svg?max-width=637&scale=1.18182) . All phenothiazine systems 8a–d are excited at longer wavelengths, i.e., at lower energies, than the carbazole dyads 8e and 8f. Eventually, the absorption characteristics of phenothiazine dyads can be more easily red-shifted and, therefore, charge separation by PET should be accessible with visible light by fine-tuning the donor chromophore towards lower HOMO–LUMO gaps.
. All phenothiazine systems 8a–d are excited at longer wavelengths, i.e., at lower energies, than the carbazole dyads 8e and 8f. Eventually, the absorption characteristics of phenothiazine dyads can be more easily red-shifted and, therefore, charge separation by PET should be accessible with visible light by fine-tuning the donor chromophore towards lower HOMO–LUMO gaps.
Conclusion
The Ugi four-component reaction represents a rapid and excellent modular and diversity-oriented synthesis of donor-anthraquinone dyads with various phenothiazine and carbazole model donors. Cyclic voltammetry and UV–vis spectroscopy clearly indicate an electronic decoupling of the donor and the acceptor substituents in the electronic ground state, whereas the emission of the donor moieties is efficiently quenched according to static fluorescence spectroscopy. The observed peculiar fluorescence quenching was previously studied by femtosecond transient absorption spectroscopy of a related model dyad indicating a photo-induced electron transfer (PET) process into a dark, i.e., non-emissive, charge-separated state. The Gibbs free energies of the PET into the charge-separated states are exergonic and can be quickly calculated from absorption and electrochemical data applying the Weller approximation. The concise synthetic concept to donor–acceptor systems is very general, easy to perform and readily expandable to all kinds of functional π-electron systems. The Weller approximation of the Gibbs free energies of the PET allows a semiquantitative evaluation and optimization of photo-induced charge-separation systems. Studies directed towards multicomponent syntheses of more complex light harvesting and charge separation systems are currently underway.
Experimental
Synthesis of compounds 8 and 10 via Ugi four-component reaction (General Procedure) in a manner similar to [56])
In a 25 mL Schlenk tube 0.50 mmol of the donor hydrochloride 4 and potassium hydroxide (28 mg, 0.50 mmol) were dissolved in methanol and the mixture was stirred for 30 min (for experimental details see Table 5). A solution of aldehyde 5 (118 mg, 0.50 mmol) in dichloromethane (2 mL) or the neat aldehyde 9 (22 mg, 0.50 mmol) were added to the reaction mixture and the solution was stirred at rt for 1 h, followed by the addition of 1 equiv of acetic acid (6) (30 mg, 0.50 mmol) and 1 equiv of tert-butyl isocyanide (7) (42 mg, 0.50 mmol) by syringe. The reaction mixture was stirred overnight at rt. The solvents were removed in vacuo and the crude product was purified by column chromatography on silica gel to give the analytically pure Ugi products 8 and 10.
Table 5: Experimental details for the synthesis of the Ugi products 8 and 10.
| Entry | MeOH [mL] | CH2Cl2 [mL] | Reaction time t [d] | Ugi 4CR products 8 or 10 (yield) |
|---|---|---|---|---|
| 1 | 3 | 2 | 2 | 153 mg (50 %) of 8a |
| 2 | 2 | 2 | 1 | 193 mg(57 %) of 8b |
| 3 | 2 | 2 | 1 | 299 mg(80 %) of 8c |
| 4 | 2 | 2 | 1 | 287 mg (60 %) of 8d |
| 5 | 2 | 2.2 | 1 | 187 mg (55 %) of 8e |
| 6 | 2 | 2 | 1 | 162 mg (45 %) of 8f |
| 7 | 3 | – | 1 | 105 mg (35 %) of 10a |
| 8 | 2 | – | 1 | 162 mg (72 %) of 10b |
Supporting Information
| Supporting Information File 1: 1H NMR, 13C NMR, UV–vis, fluorescence spectra and cyclic voltammograms of compounds 8 and 10, a summary of the X-ray crystallographic data of S(O)-1, computed xyz-coordinates of the structure 1 and HOMO and LUMO energies. | ||
| Format: PDF | Size: 2.4 MB | Download |
References
-
Müller, T. J. J.; Bunz, U. H. F., Eds. Functional Organic Materials. Syntheses, Strategies, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007.
Return to citation in text: [1] -
Müllen, K.; Wegner, G., Eds. Electronic Materials: The Oligomer Approach; Wiley-VCH: Weinheim, Germany, 1998.
Return to citation in text: [1] -
Müllen, K.; Scherf, U., Eds. Organic Light-Emitting Diodes – Synthesis, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2006.
Return to citation in text: [1] -
Kim, E.; Park, S. B. Chem.–Asian J. 2009, 4, 1646–1658. doi:10.1002/asia.200900102
Return to citation in text: [1] -
Cairo, C. W.; Key, J. A.; Sadek, C. M. Curr. Opin. Chem. Biol. 2010, 14, 57–63. doi:10.1016/j.cbpa.2009.09.032
Return to citation in text: [1] -
Wagenknecht, H.-A. Ann. N. Y. Acad. Sci. 2008, 1130, 122–130. doi:10.1196/annals.1430.001
Return to citation in text: [1] -
Petty, M. C.; Bryce, M. R.; Bloor, D., Eds. Introduction to Molecular Electronics; Oxford University Press: New York, USA, 1995.
Return to citation in text: [1] -
Balzani, V.; Credi, A.; Venturi, M. Molecular Devices and Machines - A Journey into the Nano World; Wiley-VCH: Weinheim, Germany, 2003.
Return to citation in text: [1] -
Lai, R. Y.; Fabrizio, E. F.; Lu, L.; Jenekhe, S. A.; Bard, A. J. J. Am. Chem. Soc. 2001, 123, 9112–9118. doi:10.1021/ja0102235
Return to citation in text: [1] -
Armstrong, N. R.; Wightman, R. M.; Gross, E. M. Annu. Rev. Phys. Chem. 2001, 52, 391–422. doi:10.1146/annurev.physchem.52.1.391
Return to citation in text: [1] -
Richter, M. M. Chem. Rev. 2004, 104, 3003–3036. doi:10.1021/cr020373d
Return to citation in text: [1] -
Dini, D. Chem. Mater. 2005, 17, 1933–1945. doi:10.1021/cm049567v
Return to citation in text: [1] -
Kulkarni, A. P.; Wu, P.-T.; Kwon, T. W.; Jenekhe, S. A. J. Phys. Chem. B 2005, 109, 19584–19594. doi:10.1021/jp0529772
Return to citation in text: [1] -
Kulkarni, A. P.; Zhu, Y.; Babel, A.; Wu, P.-T.; Jenekhe, S. A. Chem. Mater. 2008, 20, 4212–4223. doi:10.1021/cm7022136
Return to citation in text: [1] -
Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem., Int. Ed. 1998, 37, 402–428. doi:10.1002/(SICI)1521-3773(19980302)37:4<402::AID-ANIE402>3.0.CO;2-9
Return to citation in text: [1] -
Mitschke, U.; Bäuerle, P. J. Mater. Chem. 2000, 10, 1471–1507. doi:10.1039/A908713C
Return to citation in text: [1] -
Kulkarni, A. P.; Tonzola, C. J.; Babel, A.; Jenekhe, S. A. Chem. Mater. 2004, 16, 4556–4573. doi:10.1021/cm049473l
Return to citation in text: [1] -
Mishra, A.; Fischer, M. K. R.; Bäuerle, P. Angew. Chem., Int. Ed. 2009, 48, 2474–2499. doi:10.1002/anie.200804709
Return to citation in text: [1] -
Armstrong, N. R.; Wang, W.; Alloway, D. M.; Placencia, D.; Ratcliff, E.; Brumbach, M. Macromol. Rapid Commun. 2009, 30, 717–731. doi:10.1002/marc.200900075
Return to citation in text: [1] -
Linton, K. E.; Fisher, A. L.; Pearson, C.; Fox, M. A.; Pålsson, L.-O.; Bryce, M. R.; Petty, M. C. J. Mater. Chem. 2012, 22, 11816–11825. doi:10.1039/c2jm31825c
Return to citation in text: [1] -
Hoppe, H.; Sariciftci, N. S. J. Mater. Res. 2004, 19, 1924–1945. doi:10.1557/JMR.2004.0252
Return to citation in text: [1] -
Sun, X.; Liu, Y.; Xu, X.; Yang, C.; Yu, G.; Chen, S.; Zhao, Z.; Qiu, W.; Li, Y.; Zhu, D. J. Phys. Chem. B 2005, 109, 10786–10792. doi:10.1021/jp0509515
Return to citation in text: [1] [2] -
Sonar, P.; Lim, J. P. F.; Chan, K. L. Energy Environ. Sci. 2011, 4, 1558–1574. doi:10.1039/C0EE00668H
Return to citation in text: [1] -
Walker, B.; Kim, C.; Nguyen, T.-Q. Chem. Mater. 2011, 23, 470–482. doi:10.1021/cm102189g
Return to citation in text: [1] -
Lin, Y.; Li, Y.; Zhan, X. Chem. Soc. Rev. 2012, 41, 4245–4272. doi:10.1039/C2CS15313K
Return to citation in text: [1] -
McConnell, I.; Li, G.; Brudvig, G. W. Chem. Biol. 2010, 17, 434–447. doi:10.1016/j.chembiol.2010.05.005
Return to citation in text: [1] -
Alstrum-Acevedo, J. H.; Brennaman, M. K.; Meyer, T. J. Inorg. Chem. 2005, 44, 6802–6827. doi:10.1021/ic050904r
Return to citation in text: [1] -
Gust, D.; Moore, T. A.; Moore, A. L. Acc. Chem. Res. 2001, 34, 40–48. doi:10.1021/ar9801301
Return to citation in text: [1] -
Sun, L.; Hammarström, L.; Åkermark, B.; Styring, S. Chem. Soc. Rev. 2001, 30, 36–49. doi:10.1039/A801490F
Return to citation in text: [1] -
Kavarnos, G. J. Fundamentals of Photoinduced Electron Transfer; Wiley VCH: Weinheim, New York, 1993.
Return to citation in text: [1] -
Wenger, O. S. Chem. Soc. Rev. 2011, 40, 3538–3550. doi:10.1039/C1CS15044H
Return to citation in text: [1] -
Lemmetyinen, H.; Tkachenko, N. V.; Efimov, A.; Niemi, M. Phys. Chem. Chem. Phys. 2011, 13, 397–412. doi:10.1039/C0CP01106A
Return to citation in text: [1] -
Vauthey, E. ChemPhysChem 2012, 13, 2001–2011. doi:10.1002/cphc.201200106
Return to citation in text: [1] -
Ricks, A. B.; Brown, K. E.; Wenninger, M.; Karlen, S. D.; Berlin, Y. A.; Co, D. T.; Wasielewski, M. R. J. Am. Chem. Soc. 2012, 134, 4581–4588. doi:10.1021/ja205913q
Return to citation in text: [1] -
Wróbel, D.; Graja, A. Coord. Chem. Rev. 2011, 255, 2555–2577. doi:10.1016/j.ccr.2010.12.026
Return to citation in text: [1] -
Dössel, L. F.; Kamm, V.; Howard, I. A.; Laquai, F.; Pisula, W.; Feng, X.; Li, C.; Takase, M.; Kudernac, T.; De Feyter, S.; Müllen, K. J. Am. Chem. Soc. 2012, 134, 5876–5886. doi:10.1021/ja211504a
Return to citation in text: [1] -
Díaz, M. C.; Herranz, M. A.; Illescas, B. M.; Martín, N.; Godbert, N.; Bryce, M. R.; Luo, C.; Swartz, A.; Anderson, G.; Guldi, D. M. J. Org. Chem. 2003, 68, 7711–7721. doi:10.1021/jo034432e
Return to citation in text: [1] -
Sasaki, Y.; Araki, Y.; Ito, O.; Alam, M. M. Photochem. Photobiol. Sci. 2007, 6, 560–565. doi:10.1039/B617229F
Return to citation in text: [1] -
Suneesh, C. V.; Gopidas, K. R. J. Phys. Chem. C 2010, 114, 18725–18734. doi:10.1021/jp107606t
Return to citation in text: [1] -
Li, Z.; Dong, Q.; Li, Y.; Xu, B.; Deng, M.; Pei, J.; Zhang, J.; Chen, F.; Wen, S.; Gao, Y.; Tian, W. J. Mater. Chem. 2011, 21, 2159–2168. doi:10.1039/C0JM02510K
Return to citation in text: [1] -
Bucci, N.; Müller, T. J. J. Tetrahedron Lett. 2006, 47, 8329–8332. doi:10.1016/j.tetlet.2006.09.075
Return to citation in text: [1] [2] -
Bucci, N.; Müller, T. J. J. Tetrahedron Lett. 2006, 47, 8323–8327. doi:10.1016/j.tetlet.2006.09.076
Return to citation in text: [1] [2] -
Imahori, H.; Mori, Y.; Matano, Y. J. Photochem. Photobiol., C 2003, 4, 51–83. doi:10.1016/S1389-5567(03)00004-2
Return to citation in text: [1] -
Rodríguez-Morgade, M. S.; Plonska-Brzezinska, M. E.; Athans, A. J.; Carbonell, E.; de Miguel, G.; Guldi, D. M.; Echegoyen, L.; Torres, T. J. Am. Chem. Soc. 2009, 131, 10484–10496. doi:10.1021/ja902471w
Return to citation in text: [1] -
Sandanayaka, A. S. D.; Sasabe, H.; Takata, T.; Ito, O. J. Photochem. Photobiol., C 2010, 11, 73–92. doi:10.1016/j.jphotochemrev.2010.05.001
Return to citation in text: [1] -
Wasielewski, M. R. Chem. Rev. 1992, 92, 435–461. doi:10.1021/cr00011a005
Return to citation in text: [1] -
Kurreck, H.; Huber, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 849–866. doi:10.1002/anie.199508491
Return to citation in text: [1] -
Gouloumis, A.; González-Rodríguez, D.; Vázquez, P.; Torres, T.; Liu, S.; Echegoyen, L.; Ramey, J.; Hug, G. L.; Guldi, D. M. J. Am. Chem. Soc. 2006, 128, 12674–12684. doi:10.1021/ja055344+
Return to citation in text: [1] -
Wan, J.; Ferreira, A.; Xia, W.; Chow, C. H.; Takechi, K.; Kamat, P. V.; Jones, G., II; Vullev, V. I. J. Photochem. Photobiol., A: Chem. 2008, 197, 364–374. doi:10.1016/j.jphotochem.2008.01.016
Return to citation in text: [1] -
Kang, Y. K.; Iovine, P. M.; Therien, M. J. Coord. Chem. Rev. 2011, 255, 804–824. doi:10.1016/j.ccr.2010.12.011
Return to citation in text: [1] -
Hankache, J.; Wenger, O. S. Phys. Chem. Chem. Phys. 2012, 14, 2685–2692. doi:10.1039/C2CP23240E
Return to citation in text: [1] -
McCafferty, D. G.; Bishop, B. M.; Wall, C. G.; Hughes, S. G.; Mecklenburg, S. M.; Meyer, T. J.; Erickson, B. W. Tetrahedron 1995, 51, 1093–1106. doi:10.1016/0040-4020(94)01018-U
Return to citation in text: [1] -
Striplin, D. R.; Reece, S. Y.; McCafferty, D. G.; Wall, C. G.; Friesen, D. A.; Erickson, B. W.; Meyer, T. J. J. Am. Chem. Soc. 2004, 126, 5282–5291. doi:10.1021/ja0304289
Return to citation in text: [1] -
Myers, C. P.; Williams, M. E. Coord. Chem. Rev. 2010, 254, 2416–2428. doi:10.1016/j.ccr.2010.02.018
Return to citation in text: [1] -
Zhang, W.-W.; Mao, W.-L.; Hu, Y.-X.; Tian, Z.-Q.; Wang, Z.-L.; Meng, Q.-J. J. Phys. Chem. A 2009, 113, 9997–10004. doi:10.1021/jp903390v
Return to citation in text: [1] -
Bay, S.; Villnow, T.; Ryseck, G.; Rai-Constapel, V.; Gilch, P.; Müller, T. J. J. ChemPlusChem 2013, 78, 137–141. doi:10.1002/cplu.201200279
Return to citation in text: [1] [2] [3] [4] [5] -
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Return to citation in text: [1] -
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] -
Biggs-Houck, J. E.; Younai, A.; Shaw, J. T. Curr. Opin. Chem. Biol. 2010, 14, 371–382. doi:10.1016/j.cbpa.2010.03.003
Return to citation in text: [1] -
Qiu, G.; Ding, Q.; Wu, J. Chem. Soc. Rev. 2013, 42, 5257–5269. doi:10.1039/C3CS35507A
Return to citation in text: [1] -
Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r
Return to citation in text: [1] -
Müller, T. J. J. In Functional Organic Materials. Syntheses, Strategies, and Applications; Müller, T. J. J.; Bunz, U. H. F., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; pp 179–223.
Return to citation in text: [1] -
Müller, T. J. J.; D’Souza, D. M. Pure Appl. Chem. 2008, 80, 609–620. doi:10.1351/pac200880030609
Return to citation in text: [1] -
Yi, C.; Blum, C.; Liu, S.-X.; Frei, G.; Neels, A.; Stoeckli-Evans, H.; Leutwyler, S.; Decurtins, S. Tetrahedron 2008, 64, 9437–9441. doi:10.1016/j.tet.2008.07.084
Return to citation in text: [1] -
Samanta, A.; Vendrell, M.; Dasa, R.; Chang, Y.-T. Chem. Commun. 2010, 46, 7406–7408. doi:10.1039/C0CC02366C
Return to citation in text: [1] -
Vendrell, M.; Lee, J.-S.; Chang, Y.-T. Curr. Opin. Chem. Biol. 2010, 14, 383–389. doi:10.1016/j.cbpa.2010.02.020
Return to citation in text: [1] -
Main, M.; Snaith, J. S.; Meloni, M. M.; Jauregui, M.; Sykes, D.; Faulkner, S.; Kenwright, A. M. Chem. Commun. 2008, 5212–5214. doi:10.1039/B810083G
Return to citation in text: [1] -
Briehn, C. A.; Bäuerle, P. Chem. Commun. 2002, 1015–1023. doi:10.1039/B108846G
Return to citation in text: [1] -
Briehn, C. A.; Schiedel, M.-S.; Bonsen, E. M.; Schuhmann, W.; Bäuerle, P. Angew. Chem., Int. Ed. 2001, 40, 4680–4683. doi:10.1002/1521-3773(20011217)40:24<4680::AID-ANIE4680>3.0.CO;2-X
Return to citation in text: [1] -
Willy, B.; Müller, T. J. J. Curr. Org. Chem. 2009, 13, 1777–1790. doi:10.2174/138527209789630479
Return to citation in text: [1] -
Willy, B.; Müller, T. J. J. ARKIVOC 2008, No. i, 195–208.
Return to citation in text: [1] -
Müller, T. J. J. Synthesis 2012, 159–174. doi:10.1055/s-0031-1289636
Return to citation in text: [1] -
Franz, A. W.; Popa, L. N.; Müller, T. J. J. Tetrahedron Lett. 2008, 49, 3300–3303. doi:10.1016/j.tetlet.2008.03.071
Return to citation in text: [1] -
Mataranga-Popa, L. N.; Franz, A. W.; Bay, S.; Müller, T. J. J. Lett. Org. Chem. 2012, 9, 211–217. doi:10.2174/157017812800167529
Return to citation in text: [1] -
CCDC 976231 (S(O)-1) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Janiak, C. J. Chem. Soc., Dalton Trans. 2000, 3885–3896. doi:10.1039/B003010O
Return to citation in text: [1] -
Yang, X.-J.; Drepper, F.; Wu, B.; Sun, W.-H.; Haehnel, W.; Janiak, C. Dalton Trans. 2005, 256–267. doi:10.1039/B414999H
And supplementary material therein.
Return to citation in text: [1] -
Sailer, M.; Nonnenmacher, M.; Oeser, T.; Müller, T. J. J. Eur. J. Org. Chem. 2006, 423–435. doi:10.1002/ejoc.200500539
Return to citation in text: [1] -
Sailer, M.; Franz, A. W.; Müller, T. J. J. Chem.–Eur. J. 2008, 14, 2602–2614. doi:10.1002/chem.200701341
Return to citation in text: [1] -
Krämer, C. S.; Zeitler, K.; Müller, T. J. J. Tetrahedron Lett. 2001, 42, 8619–8624. doi:10.1016/S0040-4039(01)01848-2
Return to citation in text: [1] -
Gautrot, J. E.; Hodge, P.; Cupertino, D.; Helliwell, M. New J. Chem. 2007, 31, 1585–1593. doi:10.1039/B701257H
Return to citation in text: [1] -
Li, J.; Kendig, C. E.; Nesterov, E. E. J. Am. Chem. Soc. 2007, 129, 15911–15918. doi:10.1021/ja0748027
Return to citation in text: [1] -
Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304
Return to citation in text: [1] -
Parr, R. G.; Yang, W. Density-functional theory of atoms and molecules; Oxford University Press: Oxford, 1989.
Return to citation in text: [1] -
Weller, A. Z. Phys. Chem. 1982, 133, 93–98.
Return to citation in text: [1] -
Lor, M.; Viaene, L.; Pilot, R.; Fron, E.; Jordens, S.; Schweitzer, G.; Weil, T.; Müllen, K.; Verhoeven, J. W.; Van der Auweraer, M.; De Schryver, F. C. J. Phys. Chem. B 2004, 108, 10721–10731. doi:10.1021/jp0490352
Return to citation in text: [1] -
Suzuki, S.; Matsumoto, Y.; Tsubamoto, M.; Sugimura, R.; Kozaki, M.; Kimoto, K.; Iwamura, M.; Nozaki, K.; Senju, N.; Uragami, C.; Hashimoto, H.; Muramatsu, Y.; Konnoe, A.; Okada, K. Phys. Chem. Chem. Phys. 2013, 15, 8088–8094. doi:10.1039/C3CP50182E
Return to citation in text: [1] -
Berberich, M.; Würthner, F. Chem. Sci. 2012, 3, 2771–2777. doi:10.1039/C2SC20554H
Return to citation in text: [1] -
SPARTAN '08, V 1.2.0; Wavefunction Inc.: Irvine, CA, 2008.
Return to citation in text: [1]
| 73. | Franz, A. W.; Popa, L. N.; Müller, T. J. J. Tetrahedron Lett. 2008, 49, 3300–3303. doi:10.1016/j.tetlet.2008.03.071 |
| 74. | Mataranga-Popa, L. N.; Franz, A. W.; Bay, S.; Müller, T. J. J. Lett. Org. Chem. 2012, 9, 211–217. doi:10.2174/157017812800167529 |
| 75. | CCDC 976231 (S(O)-1) contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 1. | Müller, T. J. J.; Bunz, U. H. F., Eds. Functional Organic Materials. Syntheses, Strategies, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. |
| 7. | Petty, M. C.; Bryce, M. R.; Bloor, D., Eds. Introduction to Molecular Electronics; Oxford University Press: New York, USA, 1995. |
| 8. | Balzani, V.; Credi, A.; Venturi, M. Molecular Devices and Machines - A Journey into the Nano World; Wiley-VCH: Weinheim, Germany, 2003. |
| 41. | Bucci, N.; Müller, T. J. J. Tetrahedron Lett. 2006, 47, 8329–8332. doi:10.1016/j.tetlet.2006.09.075 |
| 42. | Bucci, N.; Müller, T. J. J. Tetrahedron Lett. 2006, 47, 8323–8327. doi:10.1016/j.tetlet.2006.09.076 |
| 4. | Kim, E.; Park, S. B. Chem.–Asian J. 2009, 4, 1646–1658. doi:10.1002/asia.200900102 |
| 5. | Cairo, C. W.; Key, J. A.; Sadek, C. M. Curr. Opin. Chem. Biol. 2010, 14, 57–63. doi:10.1016/j.cbpa.2009.09.032 |
| 6. | Wagenknecht, H.-A. Ann. N. Y. Acad. Sci. 2008, 1130, 122–130. doi:10.1196/annals.1430.001 |
| 43. | Imahori, H.; Mori, Y.; Matano, Y. J. Photochem. Photobiol., C 2003, 4, 51–83. doi:10.1016/S1389-5567(03)00004-2 |
| 44. | Rodríguez-Morgade, M. S.; Plonska-Brzezinska, M. E.; Athans, A. J.; Carbonell, E.; de Miguel, G.; Guldi, D. M.; Echegoyen, L.; Torres, T. J. Am. Chem. Soc. 2009, 131, 10484–10496. doi:10.1021/ja902471w |
| 45. | Sandanayaka, A. S. D.; Sasabe, H.; Takata, T.; Ito, O. J. Photochem. Photobiol., C 2010, 11, 73–92. doi:10.1016/j.jphotochemrev.2010.05.001 |
| 88. | Lor, M.; Viaene, L.; Pilot, R.; Fron, E.; Jordens, S.; Schweitzer, G.; Weil, T.; Müllen, K.; Verhoeven, J. W.; Van der Auweraer, M.; De Schryver, F. C. J. Phys. Chem. B 2004, 108, 10721–10731. doi:10.1021/jp0490352 |
| 89. | Suzuki, S.; Matsumoto, Y.; Tsubamoto, M.; Sugimura, R.; Kozaki, M.; Kimoto, K.; Iwamura, M.; Nozaki, K.; Senju, N.; Uragami, C.; Hashimoto, H.; Muramatsu, Y.; Konnoe, A.; Okada, K. Phys. Chem. Chem. Phys. 2013, 15, 8088–8094. doi:10.1039/C3CP50182E |
| 3. | Müllen, K.; Scherf, U., Eds. Organic Light-Emitting Diodes – Synthesis, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2006. |
| 37. | Díaz, M. C.; Herranz, M. A.; Illescas, B. M.; Martín, N.; Godbert, N.; Bryce, M. R.; Luo, C.; Swartz, A.; Anderson, G.; Guldi, D. M. J. Org. Chem. 2003, 68, 7711–7721. doi:10.1021/jo034432e |
| 83. | Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009. |
| 84. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 85. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 86. | Parr, R. G.; Yang, W. Density-functional theory of atoms and molecules; Oxford University Press: Oxford, 1989. |
| 2. | Müllen, K.; Wegner, G., Eds. Electronic Materials: The Oligomer Approach; Wiley-VCH: Weinheim, Germany, 1998. |
| 22. | Sun, X.; Liu, Y.; Xu, X.; Yang, C.; Yu, G.; Chen, S.; Zhao, Z.; Qiu, W.; Li, Y.; Zhu, D. J. Phys. Chem. B 2005, 109, 10786–10792. doi:10.1021/jp0509515 |
| 38. | Sasaki, Y.; Araki, Y.; Ito, O.; Alam, M. M. Photochem. Photobiol. Sci. 2007, 6, 560–565. doi:10.1039/B617229F |
| 39. | Suneesh, C. V.; Gopidas, K. R. J. Phys. Chem. C 2010, 114, 18725–18734. doi:10.1021/jp107606t |
| 40. | Li, Z.; Dong, Q.; Li, Y.; Xu, B.; Deng, M.; Pei, J.; Zhang, J.; Chen, F.; Wen, S.; Gao, Y.; Tian, W. J. Mater. Chem. 2011, 21, 2159–2168. doi:10.1039/C0JM02510K |
| 56. | Bay, S.; Villnow, T.; Ryseck, G.; Rai-Constapel, V.; Gilch, P.; Müller, T. J. J. ChemPlusChem 2013, 78, 137–141. doi:10.1002/cplu.201200279 |
| 26. | McConnell, I.; Li, G.; Brudvig, G. W. Chem. Biol. 2010, 17, 434–447. doi:10.1016/j.chembiol.2010.05.005 |
| 27. | Alstrum-Acevedo, J. H.; Brennaman, M. K.; Meyer, T. J. Inorg. Chem. 2005, 44, 6802–6827. doi:10.1021/ic050904r |
| 30. | Kavarnos, G. J. Fundamentals of Photoinduced Electron Transfer; Wiley VCH: Weinheim, New York, 1993. |
| 31. | Wenger, O. S. Chem. Soc. Rev. 2011, 40, 3538–3550. doi:10.1039/C1CS15044H |
| 32. | Lemmetyinen, H.; Tkachenko, N. V.; Efimov, A.; Niemi, M. Phys. Chem. Chem. Phys. 2011, 13, 397–412. doi:10.1039/C0CP01106A |
| 33. | Vauthey, E. ChemPhysChem 2012, 13, 2001–2011. doi:10.1002/cphc.201200106 |
| 34. | Ricks, A. B.; Brown, K. E.; Wenninger, M.; Karlen, S. D.; Berlin, Y. A.; Co, D. T.; Wasielewski, M. R. J. Am. Chem. Soc. 2012, 134, 4581–4588. doi:10.1021/ja205913q |
| 79. | Sailer, M.; Franz, A. W.; Müller, T. J. J. Chem.–Eur. J. 2008, 14, 2602–2614. doi:10.1002/chem.200701341 |
| 80. | Krämer, C. S.; Zeitler, K.; Müller, T. J. J. Tetrahedron Lett. 2001, 42, 8619–8624. doi:10.1016/S0040-4039(01)01848-2 |
| 21. | Hoppe, H.; Sariciftci, N. S. J. Mater. Res. 2004, 19, 1924–1945. doi:10.1557/JMR.2004.0252 |
| 22. | Sun, X.; Liu, Y.; Xu, X.; Yang, C.; Yu, G.; Chen, S.; Zhao, Z.; Qiu, W.; Li, Y.; Zhu, D. J. Phys. Chem. B 2005, 109, 10786–10792. doi:10.1021/jp0509515 |
| 23. | Sonar, P.; Lim, J. P. F.; Chan, K. L. Energy Environ. Sci. 2011, 4, 1558–1574. doi:10.1039/C0EE00668H |
| 24. | Walker, B.; Kim, C.; Nguyen, T.-Q. Chem. Mater. 2011, 23, 470–482. doi:10.1021/cm102189g |
| 25. | Lin, Y.; Li, Y.; Zhan, X. Chem. Soc. Rev. 2012, 41, 4245–4272. doi:10.1039/C2CS15313K |
| 35. | Wróbel, D.; Graja, A. Coord. Chem. Rev. 2011, 255, 2555–2577. doi:10.1016/j.ccr.2010.12.026 |
| 36. | Dössel, L. F.; Kamm, V.; Howard, I. A.; Laquai, F.; Pisula, W.; Feng, X.; Li, C.; Takase, M.; Kudernac, T.; De Feyter, S.; Müllen, K. J. Am. Chem. Soc. 2012, 134, 5876–5886. doi:10.1021/ja211504a |
| 81. | Gautrot, J. E.; Hodge, P.; Cupertino, D.; Helliwell, M. New J. Chem. 2007, 31, 1585–1593. doi:10.1039/B701257H |
| 82. | Li, J.; Kendig, C. E.; Nesterov, E. E. J. Am. Chem. Soc. 2007, 129, 15911–15918. doi:10.1021/ja0748027 |
| 15. | Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem., Int. Ed. 1998, 37, 402–428. doi:10.1002/(SICI)1521-3773(19980302)37:4<402::AID-ANIE402>3.0.CO;2-9 |
| 16. | Mitschke, U.; Bäuerle, P. J. Mater. Chem. 2000, 10, 1471–1507. doi:10.1039/A908713C |
| 17. | Kulkarni, A. P.; Tonzola, C. J.; Babel, A.; Jenekhe, S. A. Chem. Mater. 2004, 16, 4556–4573. doi:10.1021/cm049473l |
| 18. | Mishra, A.; Fischer, M. K. R.; Bäuerle, P. Angew. Chem., Int. Ed. 2009, 48, 2474–2499. doi:10.1002/anie.200804709 |
| 19. | Armstrong, N. R.; Wang, W.; Alloway, D. M.; Placencia, D.; Ratcliff, E.; Brumbach, M. Macromol. Rapid Commun. 2009, 30, 717–731. doi:10.1002/marc.200900075 |
| 20. | Linton, K. E.; Fisher, A. L.; Pearson, C.; Fox, M. A.; Pålsson, L.-O.; Bryce, M. R.; Petty, M. C. J. Mater. Chem. 2012, 22, 11816–11825. doi:10.1039/c2jm31825c |
| 76. | Janiak, C. J. Chem. Soc., Dalton Trans. 2000, 3885–3896. doi:10.1039/B003010O |
| 77. |
Yang, X.-J.; Drepper, F.; Wu, B.; Sun, W.-H.; Haehnel, W.; Janiak, C. Dalton Trans. 2005, 256–267. doi:10.1039/B414999H
And supplementary material therein. |
| 9. | Lai, R. Y.; Fabrizio, E. F.; Lu, L.; Jenekhe, S. A.; Bard, A. J. J. Am. Chem. Soc. 2001, 123, 9112–9118. doi:10.1021/ja0102235 |
| 10. | Armstrong, N. R.; Wightman, R. M.; Gross, E. M. Annu. Rev. Phys. Chem. 2001, 52, 391–422. doi:10.1146/annurev.physchem.52.1.391 |
| 11. | Richter, M. M. Chem. Rev. 2004, 104, 3003–3036. doi:10.1021/cr020373d |
| 12. | Dini, D. Chem. Mater. 2005, 17, 1933–1945. doi:10.1021/cm049567v |
| 13. | Kulkarni, A. P.; Wu, P.-T.; Kwon, T. W.; Jenekhe, S. A. J. Phys. Chem. B 2005, 109, 19584–19594. doi:10.1021/jp0529772 |
| 14. | Kulkarni, A. P.; Zhu, Y.; Babel, A.; Wu, P.-T.; Jenekhe, S. A. Chem. Mater. 2008, 20, 4212–4223. doi:10.1021/cm7022136 |
| 28. | Gust, D.; Moore, T. A.; Moore, A. L. Acc. Chem. Res. 2001, 34, 40–48. doi:10.1021/ar9801301 |
| 29. | Sun, L.; Hammarström, L.; Åkermark, B.; Styring, S. Chem. Soc. Rev. 2001, 30, 36–49. doi:10.1039/A801490F |
| 41. | Bucci, N.; Müller, T. J. J. Tetrahedron Lett. 2006, 47, 8329–8332. doi:10.1016/j.tetlet.2006.09.075 |
| 42. | Bucci, N.; Müller, T. J. J. Tetrahedron Lett. 2006, 47, 8323–8327. doi:10.1016/j.tetlet.2006.09.076 |
| 78. | Sailer, M.; Nonnenmacher, M.; Oeser, T.; Müller, T. J. J. Eur. J. Org. Chem. 2006, 423–435. doi:10.1002/ejoc.200500539 |
| 55. | Zhang, W.-W.; Mao, W.-L.; Hu, Y.-X.; Tian, Z.-Q.; Wang, Z.-L.; Meng, Q.-J. J. Phys. Chem. A 2009, 113, 9997–10004. doi:10.1021/jp903390v |
| 46. | Wasielewski, M. R. Chem. Rev. 1992, 92, 435–461. doi:10.1021/cr00011a005 |
| 47. | Kurreck, H.; Huber, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 849–866. doi:10.1002/anie.199508491 |
| 48. | Gouloumis, A.; González-Rodríguez, D.; Vázquez, P.; Torres, T.; Liu, S.; Echegoyen, L.; Ramey, J.; Hug, G. L.; Guldi, D. M. J. Am. Chem. Soc. 2006, 128, 12674–12684. doi:10.1021/ja055344+ |
| 49. | Wan, J.; Ferreira, A.; Xia, W.; Chow, C. H.; Takechi, K.; Kamat, P. V.; Jones, G., II; Vullev, V. I. J. Photochem. Photobiol., A: Chem. 2008, 197, 364–374. doi:10.1016/j.jphotochem.2008.01.016 |
| 50. | Kang, Y. K.; Iovine, P. M.; Therien, M. J. Coord. Chem. Rev. 2011, 255, 804–824. doi:10.1016/j.ccr.2010.12.011 |
| 51. | Hankache, J.; Wenger, O. S. Phys. Chem. Chem. Phys. 2012, 14, 2685–2692. doi:10.1039/C2CP23240E |
| 90. | Berberich, M.; Würthner, F. Chem. Sci. 2012, 3, 2771–2777. doi:10.1039/C2SC20554H |
| 52. | McCafferty, D. G.; Bishop, B. M.; Wall, C. G.; Hughes, S. G.; Mecklenburg, S. M.; Meyer, T. J.; Erickson, B. W. Tetrahedron 1995, 51, 1093–1106. doi:10.1016/0040-4020(94)01018-U |
| 53. | Striplin, D. R.; Reece, S. Y.; McCafferty, D. G.; Wall, C. G.; Friesen, D. A.; Erickson, B. W.; Meyer, T. J. J. Am. Chem. Soc. 2004, 126, 5282–5291. doi:10.1021/ja0304289 |
| 54. | Myers, C. P.; Williams, M. E. Coord. Chem. Rev. 2010, 254, 2416–2428. doi:10.1016/j.ccr.2010.02.018 |
| 56. | Bay, S.; Villnow, T.; Ryseck, G.; Rai-Constapel, V.; Gilch, P.; Müller, T. J. J. ChemPlusChem 2013, 78, 137–141. doi:10.1002/cplu.201200279 |
| 56. | Bay, S.; Villnow, T.; Ryseck, G.; Rai-Constapel, V.; Gilch, P.; Müller, T. J. J. ChemPlusChem 2013, 78, 137–141. doi:10.1002/cplu.201200279 |
| 62. | Müller, T. J. J. In Functional Organic Materials. Syntheses, Strategies, and Applications; Müller, T. J. J.; Bunz, U. H. F., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; pp 179–223. |
| 63. | Müller, T. J. J.; D’Souza, D. M. Pure Appl. Chem. 2008, 80, 609–620. doi:10.1351/pac200880030609 |
| 64. | Yi, C.; Blum, C.; Liu, S.-X.; Frei, G.; Neels, A.; Stoeckli-Evans, H.; Leutwyler, S.; Decurtins, S. Tetrahedron 2008, 64, 9437–9441. doi:10.1016/j.tet.2008.07.084 |
| 65. | Samanta, A.; Vendrell, M.; Dasa, R.; Chang, Y.-T. Chem. Commun. 2010, 46, 7406–7408. doi:10.1039/C0CC02366C |
| 66. | Vendrell, M.; Lee, J.-S.; Chang, Y.-T. Curr. Opin. Chem. Biol. 2010, 14, 383–389. doi:10.1016/j.cbpa.2010.02.020 |
| 67. | Main, M.; Snaith, J. S.; Meloni, M. M.; Jauregui, M.; Sykes, D.; Faulkner, S.; Kenwright, A. M. Chem. Commun. 2008, 5212–5214. doi:10.1039/B810083G |
| 68. | Briehn, C. A.; Bäuerle, P. Chem. Commun. 2002, 1015–1023. doi:10.1039/B108846G |
| 69. | Briehn, C. A.; Schiedel, M.-S.; Bonsen, E. M.; Schuhmann, W.; Bäuerle, P. Angew. Chem., Int. Ed. 2001, 40, 4680–4683. doi:10.1002/1521-3773(20011217)40:24<4680::AID-ANIE4680>3.0.CO;2-X |
| 70. | Willy, B.; Müller, T. J. J. Curr. Org. Chem. 2009, 13, 1777–1790. doi:10.2174/138527209789630479 |
| 71. | Willy, B.; Müller, T. J. J. ARKIVOC 2008, No. i, 195–208. |
| 61. | Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r |
| 56. | Bay, S.; Villnow, T.; Ryseck, G.; Rai-Constapel, V.; Gilch, P.; Müller, T. J. J. ChemPlusChem 2013, 78, 137–141. doi:10.1002/cplu.201200279 |
| 56. | Bay, S.; Villnow, T.; Ryseck, G.; Rai-Constapel, V.; Gilch, P.; Müller, T. J. J. ChemPlusChem 2013, 78, 137–141. doi:10.1002/cplu.201200279 |
| 57. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
| 58. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 59. | Biggs-Houck, J. E.; Younai, A.; Shaw, J. T. Curr. Opin. Chem. Biol. 2010, 14, 371–382. doi:10.1016/j.cbpa.2010.03.003 |
| 60. | Qiu, G.; Ding, Q.; Wu, J. Chem. Soc. Rev. 2013, 42, 5257–5269. doi:10.1039/C3CS35507A |
© 2014 Bay et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)