Abstract
A novel approach for the catalyzed formation of bisindolylmethane derivatives (BIMs) is described. This methodology is the unique example where AgOTf has been successfully used for the activation of aldehydes, giving easy access to a broad range of bisindolyl derivatives with excellent results. Moreover, the simplicity and easy operational methodology using a small amount of commercially available AgOTf (1–3 mol %), one of the lowest catalytic charge used in this process to date, makes this procedure an alternative approach for this interesting and appealing reaction.

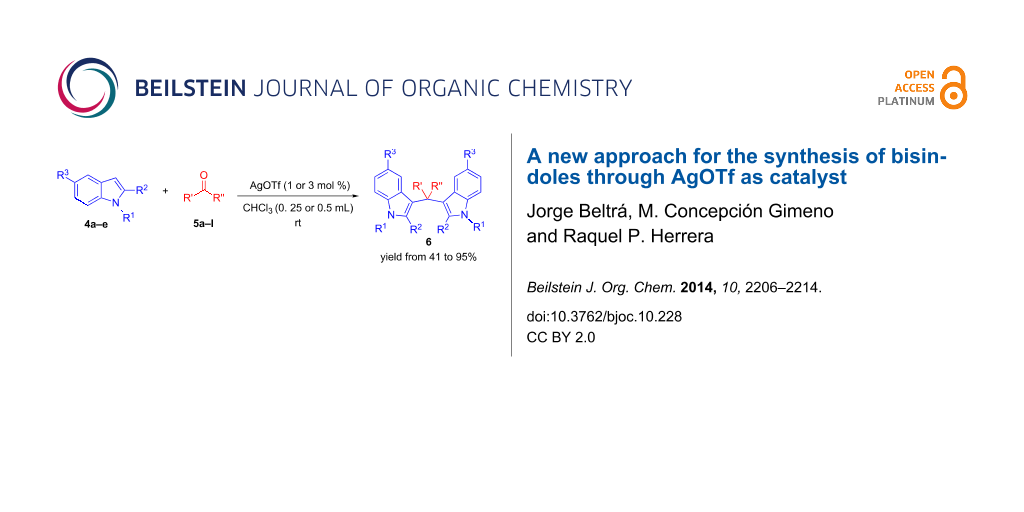
Graphical Abstract
Introduction
Indole is an interesting structural motif present in more than 3000 isolated natural products and embedded in many biological systems [1-3]. Although this field has attracted a considerable attention over the past decades, the development of new synthetic [4,5] and catalytic [6-11] methods leading to functionalized indole derivatives is still an active field because their application in drug discovery [12-14]. Indole is the structural core unit of bisindolylmethane derivatives (BIMs), many of them isolated from marine natural sources [15,16] and are an important class of indole derivatives which exhibit an appealing range of biological properties such as antibacterial, antifungal, antimicrobial or anti-inflammatory, among others (Figure 1) [17]. Interestingly, BIMs have also been found to be potent anticancer derivatives, also having antimetastatic activity, and clinical studies have demonstrated that they could be potentially used as chemotherapeutic agents against numerous forms of cancer [18-20].
![[1860-5397-10-228-1]](/bjoc/content/figures/1860-5397-10-228-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bisindolyl based important targets: 1 [21], 2 [22] and 3 [23].
Figure 1: Bisindolyl based important targets: 1 [21], 2 [22] and 3 [23].
The great importance of bisindolylmethanes, has led many efforts dedicated to the development of new strategies for their synthesis after the pioneering work reported by Fischer in 1886 [24,25], and more particularly when aldehydes are involved in their synthesis [26]. Considering the broad scope of application of BIMs together with their interesting biological properties, the development of new efficient, general and eco-friendly protocols using benign catalytic systems with structurally different aldehydes and easy accessible catalysts still remains a continuous demand.
Among the variety of Lewis acids employed in this reaction such as In(OTf)3 (5 mol %)/[omim][PF6−] [27], LiClO4 (10 equiv) [28], Cp2ZrCl2 (5 mol %) [29], Yb-amberlyst (16 mol %) [30], SmI2(THF)2 (10 mol %) [31], Sb2(SO4)3 (5 mol %) [32], Dy(OTf)3 (5–10 mol %) [33], Ln(OTf)3 (0.10 M) [34], FeCl3·6H2O (5 mol %)/[omim][PF6−] [35], V(HSO4)3 (10 mol %) [36], Bi(NO3)3 (2–5 mol %) [37], or CeCl3·7H2O (10 M) [38], Ag is a good alternative since it is one of the most abundant, cheaper, readily available metals and also displays low toxicity. We disclose here a pioneering strategy for the synthesis of bisindolylmethane derivatives via Ag activation of aldehydes. To the best of our knowledge, this is the first example where catalytic amounts of AgOTf afford the desired bisindolyl products [39,40]. Our approach could significantly broaden the scope of this reaction, providing a new and convenient protocol and avoiding some reported drawbacks, such as the use of toxic metal ions, expensive solvents, long reaction times, high temperature, anhydrous conditions, tedious work-up, low product yields, higher catalyst loading, reaction limited to aromatic aldehydes and formation of large amounts of wastes or byproducts.
Results and Discussion
In the context of our research program focused on the synthesis of new indole derivatives, we centered our attention in the preparation of bisindolylmethane derivatives following a very easy and straightforward procedure, in order to contribute to the development of this field. Accordingly, we decided to evaluate the possibility of preparing BIMs from aldehydes and simple Ag(I) species (Scheme 1).
![[1860-5397-10-228-i1]](/bjoc/content/inline/1860-5397-10-228-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Test reaction using diverse Ag(I) species.
Scheme 1: Test reaction using diverse Ag(I) species.
In order to test the viability of this idea, the investigation was started by exploring the efficiency of four easily accessible Ag(I) species in two reaction models (Scheme 1), and following the course of the process by TLC until no qualitative advance of the reaction was observed. To our delight, AgOTf allowed an almost full conversion in a very clean reaction after less than 12 hours, measuring the conversion of the process by 1H NMR. In contrast, AgNO3, Ag2CO3 and AgOAc showed a lack of reactivity for both aldehydes 5a and 5b. This could be in agreement with the higher coordination capacity of the tested counterions, while OTf is considered to be low-coordinating with some cations, thus favouring the Lewis acid character of Ag(I) [41,42]. Bearing this promising result in mind, the ensuing screening was performed using AgOTf as catalyst and exploring different key parameters of the reaction in order to reach the best reaction conditions (Table 1).
Table 1: Screening of the catalytic synthesis of BIM derivatives 6aa.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-228-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent (mL) | AgOTf (mol %) | Time (h) | Conv. (%)b |
|---|---|---|---|---|
| 1 | Toluene (0.5) | 5 | 3.5 | 84 |
| 2 | CH3CN (0.5) | 5 | 3.5 | 84 |
| 3 | THF (0.5) | 5 | 3.5 | 89 |
| 4 | CHCl3 (0.5) | 5 | 3.5 | 93 |
| 5 | EtOAc (0.5) | 5 | 3.5 | 78 |
| 6 | MeOH (0.5) | 5 | 3.5 | <5 |
| 7 | CHCl3 (0.5) | 3 | 5 | 64 |
| 8 | CHCl3 (0.5) | 3 | 18 | 83 |
| 9c | CHCl3 (0.5) | 3 | 18 | 88 |
| 10d | CHCl3 (0.5) | 3 | 18 | 83 |
| 11d | CHCl3 (0.25) | 3 | 18 | 79 |
| 12d | CHCl3 (0.5) | 1 | 24 | 89 |
| 13d | CHCl3 (0.25) | 1 | 24 | 86 |
| 14c,d | CHCl3 (0.5) | 1 | 24 | 81 |
| 15 | CHCl3 (0.5) | – | 24 | <5 |
aExperimental conditions: AgOTf (1 or 3 mol %), indole (4a, 1 mmol) in 0.25 or 0.5 mL of CHCl3 and aldehyde 5a (0.5 mmol). bConversion calculated by 1H NMR using dimethyl fumarate as internal standard. cReaction performed using 1.5 equiv of indole. dReaction performed for 0.5 mmol scale of aldehyde.
In the first screening of solvent, encouraging results were achieved with CHCl3 for 5 mol % of catalyst (Table 1, entry 4) in short reactions times (5 h), without heating the reaction. Good results were also obtained for the rest of solvents tested, with the exception of MeOH which exhibited almost no reactivity (Table 1, entry 6). At this point, and because there is a big concern about sustainable chemistry, the next step was centered in decreasing the catalyst loading without reducing the yield of the process, although longer reaction time was required. Interestingly, it was possible to decrease the catalytic amount of the catalyst until 1 mol %, to the best of our knowledge one of the smallest catalyst loading used in this reaction to date (Table 1, entry 12), although longer reaction times were necessary compared with the use of 3 mol % (Table 1, entry 8). In all cases, very clean crude products were found at the end of the process. At this point, the amount of aldehyde was scaled up to 0.5 mmol in order to facilitate the weighing of the catalyst without decreasing the yield of the process (Table 1, entries 10–14). Surprisingly, variation in the dilution of the reaction afforded similar yield for the same reaction time (compare Table 1, entries 10 and 11 and entries 12 and 13). On the other hand, the addition of an excess of indole did not give rise much better results (Table 1, entries 9 and 14). In order to prove the efficiency of this catalytic system the background of the process was tested, demonstrating a lack of reactivity in absence of the catalyst (Table 1, entry 15). With all these results in mind, the scope of the reaction was continued with 1 or 3 mol % of catalyst in CHCl3 (0.25–0.50 mL) (Table 2) as the best reaction conditions.
Table 2: Scope of the catalytic synthesis of BIM derivatives 6.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-228-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Indole | Aldehyde | Solvent (mL) | Cat. (mol %) | Time (h) | Product | Yield (%)b |
|---|---|---|---|---|---|---|---|
| 1c |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-228-i6.svg?max-width=637&scale=1.0)
4a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-228-i7.svg?max-width=637&scale=1.0)
5a |
5 | 1 | 18 | 6aa | >95 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-228-i8.svg?max-width=637&scale=1.0)
4a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-228-i9.svg?max-width=637&scale=1.0)
5b |
0.25 | 1 | 18 | 6ab | 89 |
| 3d |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-228-i10.svg?max-width=637&scale=1.0)
4a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-228-i11.svg?max-width=637&scale=1.0)
5c |
0.50 | 1 | 15 | 6ac | 90 |
| 4d |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-228-i12.svg?max-width=637&scale=1.0)
4a |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-228-i13.svg?max-width=637&scale=1.0)
5d |
0.50 | 5 | 48 | 6ad | 89 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-228-i14.svg?max-width=637&scale=1.0)
4a |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-228-i15.svg?max-width=637&scale=1.0)
5e |
0.25 | 3 | 18 | 6ae | 81 |
| 6d |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-228-i16.svg?max-width=637&scale=1.0)
4a |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-228-i17.svg?max-width=637&scale=1.0)
5f |
0.50 | 1 | 16 | 6af | >95 |
| 7d |
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-228-i18.svg?max-width=637&scale=1.0)
4a |
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-228-i19.svg?max-width=637&scale=1.0)
5g |
0.50 | 3 | 24 | 6ag | 41 |
| 8d |
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-228-i20.svg?max-width=637&scale=1.0)
4a |
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-228-i21.svg?max-width=637&scale=1.0)
5h |
0.50 | 1 | 18 | 6ah | 90 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-228-i22.svg?max-width=637&scale=1.0)
4a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-228-i23.svg?max-width=637&scale=1.0)
5i |
0.50 | 3 | 18 | 6ai | 95 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-228-i24.svg?max-width=637&scale=1.0)
4a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-228-i25.svg?max-width=637&scale=1.0)
5j |
0.50 | 3 | 24 | 6aj | 90 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-228-i26.svg?max-width=637&scale=1.0)
4a |
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-228-i27.svg?max-width=637&scale=1.0)
5k |
0.50 | 5 | 24 | 6ak | 70 |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-10-228-i28.svg?max-width=637&scale=1.0)
4b |
![[Graphic 26]](/bjoc/content/inline/1860-5397-10-228-i29.svg?max-width=637&scale=1.0)
5a |
0.50 | 1 | 18 | 6ba | 85 |
| 13 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-10-228-i30.svg?max-width=637&scale=1.0)
4c |
![[Graphic 28]](/bjoc/content/inline/1860-5397-10-228-i31.svg?max-width=637&scale=1.0)
5a |
0.50 | 1 | 18 | 6ca | 95 |
| 14d |
![[Graphic 29]](/bjoc/content/inline/1860-5397-10-228-i32.svg?max-width=637&scale=1.0)
4d |
![[Graphic 30]](/bjoc/content/inline/1860-5397-10-228-i33.svg?max-width=637&scale=1.0)
5a |
0.50 | 3 | 24 | 6da | 75 |
| 15d |
![[Graphic 31]](/bjoc/content/inline/1860-5397-10-228-i34.svg?max-width=637&scale=1.0)
4e |
![[Graphic 32]](/bjoc/content/inline/1860-5397-10-228-i35.svg?max-width=637&scale=1.0)
5a |
0.50 | 1 | 8 | 6ea | 93 |
aOtherwise indicated: AgOTf (1 or 3 mol %), indole 4a–e (1 mmol), CHCl3 (0.25 mL) and aldehyde/ketone 5a–k (0.5 mmol). bIsolated yields by flash chromatography (SiO2; hexane/EtOAc, 8:2). cReaction performed using 5 mmol of aldehyde 5a and 10 mmol of indole (4a), for 1.4 g scale. dFor 0.2 mmol of aldehyde.
The evaluation of the scope of this Ag(I)-catalyzed methodology was performed testing diverse commercially available aldehydes and indoles giving easy access to a great number of different substituted bisindoles 6 with very good results (Table 2).
As shown above, high yields were achieved in reasonable reaction time using a representative spectrum of aliphatic and aromatic aldehydes (Table 2, entry 1–10). Aldehyde 5d reacted slowly maybe due to steric factors (3 mol % of catalyst affords 34% yield after 24 h). However, in order to increase the reaction rate preventing the evaporation of 5d, the process was carried out with 5 mol % of catalyst and a higher yield was achieved (Table 2, entry 4). It is remarkable that the resulting product starting from aldehyde 5g afforded interesting trisindole analogue 6ag (Table 2, entry 7). Trisindoles are a less explored class of indole derivatives but with potential anticancer properties [26], and their synthesis is also an appealing challenge. Due to the high insolubility of aldehyde 5g in the reaction medium, the reaction was performed with 5 mol % of catalyst, however moderate results were obtained in this case. Different solvents were also tested in order to improve the yield of final 6ag but only a 35% yield was obtained in EtOAc and no reaction in THF, after 2 days. The use of different substituted indoles 4b–e allowed the desired products to be obtained with excellent results (Table 2, entries 12–15). There is a clear dependence between the electronic environment of the indole and the yield of the final product, since less reactivity was exhibited with indole 4d (Table 2, entry 14). Given the possibility that product 6ca could decompose in the column and in order to prevent this drawback and the decomposition of other additional products, the reaction crude was filtered through neutral alumina with Et2O, and very clean crude product spectrum was obtained. It is also possible that in some cases the yield could depend on the stability of the starting aldehyde since they are used without previous distillation. This methodology was also tested with the less active and explored ketone 5k and the reaction rendered a high yield, although 5 mol % of catalyst was required in order to decrease the reaction rate (Table 2, entry 11). Additionally, the possibility of scaling up this process was explored to obtain 1.4 g of product 6ab (Table 2, entry 2), and successfully completed conversion was achieved after 18 h of reaction with a very clean spectrum (see Supporting Information File 1).
In order to prove the applicability of this methodology, the scope of the reaction was extended to the synthesis of the interesting compound vibrindole A (Scheme 2), an isolated metabolite of the marine bacteria Vibrio parahaemolyticus, which is active against Bacillus subtilis, Staphylococcus aureus and Staphylococcus albus as has been previously demonstrated [43].
![[1860-5397-10-228-i2]](/bjoc/content/inline/1860-5397-10-228-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of biologically active vibrindole A.
Scheme 2: Synthesis of biologically active vibrindole A.
In order to prevent the loss of acetaldehyde by evaporation (bp 20 °C) and the consequent poorer yield of the final product, instead of 1 equivalent of aldehyde (0.5 mmol) 2 equivalents (1 mmol) were used. The result was 90% of yield compared with 67% for 1 equivalent of acetaldehyde, confirming our concern. It is worth mentioning that this procedure allowed excellent results using 1 mol % of catalyst, one of the lowest catalytic charges used to date. This synthesis represents an easier approach in comparison with previously reported methods for the synthesis of vibrindole A [44-49]. After evaporation of the solvent, a very clean crude product was obtained since acetaldehyde is also evaporated, and the final product was purified by a simple column chromatography. Moreover, we were able to obtain single crystals for compounds 6ad and 6al for the first time in the literature (Figure 2) [50].
![[1860-5397-10-228-2]](/bjoc/content/figures/1860-5397-10-228-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structures for compounds 6ad and 6al.
Figure 2: Crystal structures for compounds 6ad and 6al.
Although the mechanism is not clear at this stage, we can assume the same mechanism as that previously proposed for other Lewis acid (Scheme 3). In the course of the reaction after the first addition of one molecule of indole to the aldehyde, unstable intermediate 7 would promote the elimination of Ag(I), in the form of AgOH, to give azafulvene 8 [51,52]. Finally, the iminium 8 would undergo a further addition of a second molecule of indole to produce the final observed products 6 [53].
![[1860-5397-10-228-i3]](/bjoc/content/inline/1860-5397-10-228-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanism of the synthesis of bisindoles through AgOTf catalyst.
Scheme 3: Mechanism of the synthesis of bisindoles through AgOTf catalyst.
Since HOTf is released in the second step of the reaction, it is not possible to discard the participation of HOTf in the activation process, since the catalytic ability of these species has been previously invoked by other authors, when metal triflates are used as catalysts in other reactions [54]. Moreover, Brønsted acids have also been employed as promoters of this process [26]. We have performed a comparative study of the reaction between indole (4a) and aldehyde 5a, one with HOTf (1 mol %) and the other with AgOTf (1 mol %), showing that the reaction rate of the process catalyzed by triflic acid is higher (after 6 h the reaction is finished). This is also in agreement with the results observed by Hartwig and co-workers [54]. These findings indicate that AgOTf is the mean catalyst of the process, because although triflic acid could participate in the activation process, it would be consumed in the reaction with the produced AgOH to regenerate AgOTf.
Conclusion
In summary, we have reported for the first time an unprecedented approach of AgOTf-catalyzed addition of indoles to aldehydes, providing easy access to interesting BIMs with very good yields. This catalytic system has been proven active under mild reaction conditions and effective for a broad application in the synthesis of aliphatic and aromatic substituted bisiindolylmethane derivatives. The small amount of catalyst (1–3 mol %) used in our developed procedure is another advantage being one of the lowest catalytic charge used to date for this process. Furthermore, the simplicity and easy operational methodology makes this procedure an excellent alternative approach for these interesting and appealing reaction products, for which there is still a continuous demand.
Experimental
Purification of reaction products was carried out by flash chromatography using silica gel (0.063–0.200 mm). Analytical thin-layer chromatography was performed on 0.25 mm silical gel 60-F plates. 1H NMR spectra were recorded at 400 MHz; 13C APT-NMR spectra were recorded at 100 MHz; CDCl3 as the solvent. Chemical shifts were reported in the δ scale relative to the central line of CDCl3 (77 ppm) for 13C APT-NMR. All commercially available solvents and reagents were used as received. The 1H and 13C NMR spectra for compounds 6aa [55], 6ab [56], 6ac [57], 6ad [58], 6ae [35], 6af [59], 6ag [23], 6ah [60], 6ai [61], 6aj [58], 6ak [57], 6ba [62], 6ca [63], 6da [64], 6ea [64], are consistent with values previously reported in the literature.
Representative procedure for the synthesis of bisindoles 6: To a mixture of catalyst AgOTf (1 or 3 mol %) and indole 4a–e (1 mmol) in CHCl3 (0.25–0.5 mL), aldehyde 5a–k (0.5 mmol) was further added in a test tube at room temperature. After the reaction time (see Table 2), the crude mixture was filtered through neutral alumina with Et2O and the solvent was removed under vacuum. Then, adducts 6 were finally isolated by flash chromatography (SiO2; hexane/EtOAc, 8:2). Yield for each final product is reported in Table 2. Final products should be stored under nitrogen atmosphere and in the fridge.
3,3'-(3-phenylpropane-1,1-diyl)bis(1H-indole) (6ac)
![[Graphic 33]](/bjoc/content/inline/1860-5397-10-228-i36.svg?max-width=637&scale=1.18182)
Following the general procedure, compound 6ac was obtained after 15 h of reaction at room temperature in 90% yield. The 1H NMR spectrum is consistent with the values previously reported in [57]. 13C APT-NMR (100 MHz, CDCl3) δ 142.6 (1C), 136.6 (2C), 128.5 (2C), 128.2 (2C), 127.0 (2C), 125.6 (1C), 121.8 (2C), 121.5 (2C), 120.0 (2C), 119.6 (2C), 119.0 (2C), 111.1 (2C), 37.4 (1C), 34.4 (1C), 33.5 (1C).
3,3'-(2,2-dimethylpropane-1,1-diyl)bis(1H-indole) (6ad)
![[Graphic 34]](/bjoc/content/inline/1860-5397-10-228-i37.svg?max-width=637&scale=1.18182)
Following the general procedure, compound 6ad was obtained after 48 h of reaction at room temperature in 89% yield. The 1H NMR spectrum is consistent with the values previously reported in [59]. 13C APT-NMR (100 MHz, CDCl3) δ 135.3 (2C), 128.7 (2C), 121.9 (2C), 121.6 (2C), 119.4 (2C), 119.2 (2C), 119.1 (2C), 110.7 (2C), 43.1 (1C), 35.8 (1C), 29.0 (3C).
Supporting Information
| Supporting Information File 1: 1H NMR for all synthesized compounds, 13C APT-NMR spectra for compounds 6ac and 6ad and crystallographic data for compounds 6ad and 6al. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Ruiz-Sanchis, P.; Savina, S. A.; Albericio, F.; Álvarez, M. Chem. – Eur. J. 2011, 17, 1388–1408. doi:10.1002/chem.201001451
Return to citation in text: [1] -
Gribble, G. W. Top. Heterocycl. Chem. 2010, 26, 1–480. doi:10.1007/978-3-642-15733-2
Return to citation in text: [1] -
Vicente, R. Org. Biomol. Chem. 2011, 9, 6469–6480. doi:10.1039/C1OB05750B
And references therein cited.
Return to citation in text: [1] -
Joule, J. A. Indole and its Derivatives. In Science of Synthesis; Thomas, E. J., Ed.; Houben-Weyl Methods of Molecular Transformations, Vol. 10; Thieme: Stuttgart, 2000; chapter 10.13.
Return to citation in text: [1] -
Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875–2911. doi:10.1021/cr0505270
Return to citation in text: [1] -
Bandini, M.; Melloni, A.; Tommasi, S.; Umani-Ronchi, A. Synlett 2005, 1199–1222. doi:10.1055/s-2005-865210
Return to citation in text: [1] -
Marqués-López, E.; Diez-Martinez, A.; Merino, P.; Herrera, R. P. Curr. Org. Chem. 2009, 13, 1585–1609. doi:10.2174/138527209789578126
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608–9644. doi:10.1002/anie.200901843
Return to citation in text: [1] -
Terrasson, V.; de Figueiredo, R. M.; Campagne, J. M. Eur. J. Org. Chem. 2010, 2635–2655. doi:10.1002/ejoc.200901492
Return to citation in text: [1] -
Zeng, M.; You, S.-L. Synlett 2010, 1289–1301. doi:10.1055/s-0029-1219929
Return to citation in text: [1] -
Chauhan, P.; Chimni, S. S. RSC Adv. 2012, 2, 6117–6134. doi:10.1039/c2ra20544k
Return to citation in text: [1] -
Lounasmaa, M.; Tolvanen, A. Nat. Prod. Rep. 2000, 17, 175–191. doi:10.1039/a809402k
Return to citation in text: [1] -
Hibino, S.; Choshi, T. Nat. Prod. Rep. 2001, 18, 66–87. doi:10.1039/b004055j
Return to citation in text: [1] -
Wu, Y.-J. Top. Heterocycl. Chem. 2010, 26, 1–29. doi:10.1007/7081_2010_37
Return to citation in text: [1] -
Pindur, U.; Lemster, T. Curr. Med. Chem. 2001, 8, 1681–1698. doi:10.2174/0929867013371941
Return to citation in text: [1] -
Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489–4497. doi:10.1021/cr900211p
Return to citation in text: [1] -
Safe, S.; Papineni, S.; Chintharlapalli, S. Cancer Lett. 2008, 269, 326–338. doi:10.1016/j.canlet.2008.04.021
Return to citation in text: [1] -
Bell, M. C.; Crowley-Nowick, P.; Bradlow, H. L.; Sepkovic, D. W.; Schmidt-Grimminger, D.; Howell, P.; Mayeaux, E. J.; Tucker, A.; Turbat-Herrera, E. A.; Mathis, J. M. Gynecol. Oncol. 2000, 78, 123–129. doi:10.1006/gyno.2000.5847
Return to citation in text: [1] -
Hong, C.; Firestone, G. L.; Bjeldanes, L. F. Biochem. Pharmacol. 2002, 63, 1085–1097. doi:10.1016/S0006-2952(02)00856-0
Return to citation in text: [1] -
Le, H. T.; Schaldach, C. M.; Firestone, G. L.; Bjeldanes, L. F. J. Biol. Chem. 2003, 278, 21136–21145. doi:10.1074/jbc.M300588200
Return to citation in text: [1] -
Maciejewska, D.; Niemyjska, M.; Wolska, I.; Włostowski, M.; Rasztawicka, M. Z. Naturforsch. 2004, 59b, 1137–1142.
Return to citation in text: [1] -
Lee, C.-H.; Yao, C.-F.; Huang, S.-M.; Ko, S.; Tan, Y.-H.; Lee-Chen, G.-J.; Wang, Y.-C. Cancer 2008, 113, 815–825. doi:10.1002/cncr.23619
Return to citation in text: [1] -
Veluri, R.; Oka, I.; Wagner-Döbler, I.; Laatsch, H. J. Nat. Prod. 2003, 66, 1520–1523. doi:10.1021/np030288g
Return to citation in text: [1] [2] -
Fischer, E. Chem. Ber. 1886, 19, 2988–2991. doi:10.1002/cber.188601902296
Return to citation in text: [1] -
Fischer, E. Justus Liebigs Ann. Chem. 1887, 242, 372–383. doi:10.1002/jlac.18872420311
Return to citation in text: [1] -
Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Chem. Rev. 2010, 110, 2250–2293. doi:10.1021/cr900195a
Return to citation in text: [1] [2] [3] -
Ji, S.-J.; Zhou, M.-F.; Gu, D.-G.; Wang, S.-Y.; Loh, T.-P. Synlett 2003, 2077–2079. doi:10.1055/s-2003-41464
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Murthy, C. V. S. R.; Kumar, G. M.; Madan, C. Synthesis 2001, 783–787. doi:10.1055/s-2001-12777
Return to citation in text: [1] -
Kantam, M. L.; Aziz, K.; Likhar, P. R. Catal. Lett. 2004, 98, 117–121. doi:10.1007/s10562-004-7925-5
Return to citation in text: [1] -
Yu, L.; Chen, D.; Li, J.; Wang, P. G. J. Org. Chem. 1997, 62, 3575–3581. doi:10.1021/jo961933+
Return to citation in text: [1] -
Soueidan, M.; Collin, J.; Gil, R. Tetrahedron Lett. 2006, 47, 5467–5470. doi:10.1016/j.tetlet.2006.05.169
Return to citation in text: [1] -
Srinivasa, A.; Varma, P. P.; Hulikal, V.; Mahadevan, K. M. Monatsh. Chem. 2008, 139, 111–115. doi:10.1007/s00706-007-0697-z
Return to citation in text: [1] -
Mi, X.; Luo, S.; He, J.; Cheng, J.-P. Tetrahedron Lett. 2004, 45, 4567–4570. doi:10.1016/j.tetlet.2004.04.039
Return to citation in text: [1] -
Chen, D.; Yu, L.; Wang, P. G. Tetrahedron Lett. 1996, 37, 4467–4470. doi:10.1016/0040-4039(96)00958-6
Return to citation in text: [1] -
Ji, S.-J.; Zhou, M.-F.; Gu, D.-G.; Jiang, Z.-Q.; Loh, T.-P. Eur. J. Org. Chem. 2004, 1584–1587. doi:10.1002/ejoc.200300719
Return to citation in text: [1] [2] -
Shirini, F.; Yahyazadeh, A.; Abedini, M.; Langroodi, D. I. Bull. Korean Chem. Soc. 2010, 31, 1715–1718. doi:10.5012/bkcs.2010.31.6.1715
Return to citation in text: [1] -
Khodaeia, M. M.; Ghanbary, P.; Mohammadpoor-Baltork, I.; Memarian, H. R.; Khosropour, A. R.; Nikoofar, K. J. Heterocycl. Chem. 2008, 45, 377–381. doi:10.1002/jhet.5570450213
Return to citation in text: [1] -
Silveira, C. C.; Mendes, S. R.; Líbero, F. M.; Lenardão, E. J.; Perin, G. Tetrahedron Lett. 2009, 50, 6060–6063. doi:10.1016/j.tetlet.2009.08.062
Return to citation in text: [1] -
Buscemi, G.; Biffis, A.; Tubaro, C.; Basato, M.; Graiff, C.; Tiripicchio, A. Appl. Organomet. Chem. 2010, 24, 285–290. doi:10.1002/aoc.1595
Return to citation in text: [1] -
Shaikh, A. C.; Chen, C. J. Chin. Chem. Soc. 2011, 58, 899–905. doi:10.1002/jccs.201190142
Return to citation in text: [1] -
Hashmi, A. S. K. In Silver in Organic Chemistry; Harmata, M., Ed.; Wiley: Hoboken, 2010; pp 357–379. doi:10.1002/9780470597521.ch12
Return to citation in text: [1] -
Weibel, J.-M.; Blanc, A.; Pale, P. Chem. Rev. 2008, 108, 3149–3173. doi:10.1021/cr078365q
Return to citation in text: [1] -
Bell, R.; Carmeli, S.; Sar, N. J. Nat. Prod. 1994, 57, 1587–1590. doi:10.1021/np50113a022
Return to citation in text: [1] -
Gibbs, T. J. K.; Tomkinson, N. C. O. Org. Biomol. Chem. 2005, 3, 4043–4045. doi:10.1039/b513394g
Return to citation in text: [1] -
Guo, X.; Pan, S.; Liu, J.; Li, Z. J. Org. Chem. 2009, 74, 8848–8851. doi:10.1021/jo902093p
Return to citation in text: [1] -
Niu, T.; Huang, L.; Wu, T.; Zhang, Y. Org. Biomol. Chem. 2011, 9, 273–277. doi:10.1039/c0ob00709a
Return to citation in text: [1] -
Tayebee, R.; Nehzat, F.; Rezaei-Seresht, E.; Mohammadi, F. Z.; Rafiee, E. J. Mol. Catal. A: Chem. 2011, 351, 154–164. doi:10.1016/j.molcata.2011.09.029
Return to citation in text: [1] -
Abe, T.; Nakamura, S.; Yanada, R.; Choshi, T.; Hibino, S.; Ishikura, M. Org. Lett. 2013, 15, 3622–3625. doi:10.1021/ol401486s
Return to citation in text: [1] -
Huo, C.; Kang, L.; Xu, X.; Jia, X.; Wang, X.; Xie, H.; Yuan, Y. Tetrahedron Lett. 2014, 55, 954–958. doi:10.1016/j.tetlet.2013.12.055
Return to citation in text: [1] -
CCDC-1007024 (6ad) and CCDC-1007025 (6al) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Cook, A. H.; Majer, J. R. J. Chem. Soc. 1944, 486–487. doi:10.1039/JR9440000486
Return to citation in text: [1] -
Cook, A. H.; Majer, J. R. J. Chem. Soc. 1944, 488–489. doi:10.1039/JR9440000488
Return to citation in text: [1] -
Burr, G. O.; Gortner, R. A. J. Am. Chem. Soc. 1924, 46, 1224–1246. doi:10.1021/ja01670a015
Return to citation in text: [1] -
Rosenfeld, D. C.; Shekhar, S.; Takemiya, A.; Utsunomiya, M.; Hartwig, J. F. Org. Lett. 2006, 8, 4179–4182. doi:10.1021/ol061174+
Return to citation in text: [1] [2] -
Peng, Y.-Y.; Zhang, Q.-L.; Yuan, J.-J.; Cheng, J.-P. Chin. J. Chem. 2008, 26, 2228–2232. doi:10.1002/cjoc.200890396
Return to citation in text: [1] -
Xu, H.-Y.; Zi, Y.; Xu, X.-P.; Wang, S.-Y.; Ji, S.-J. Tetrahedron 2013, 69, 1600–1605. doi:10.1016/j.tet.2012.11.098
Return to citation in text: [1] -
Ramesh, C.; Banerjee, J.; Pal, R.; Das, B. Adv. Synth. Catal. 2003, 345, 557–559. doi:10.1002/adsc.200303022
Return to citation in text: [1] [2] [3] -
An, L.-T.; Ding, F.-Q.; Zhou, J.-P.; Lu, X.-H.; Zhang, L.-L. Chin. J. Chem. 2007, 25, 822–827. doi:10.1002/cjoc.200790151
Return to citation in text: [1] [2] -
Khalafi-Nezhad, A.; Parhami, A.; Zare, A.; Moosavi Zare, A. R.; Hasaninejad, A.; Panahi, F. Synthesis 2008, 617–621. doi:10.1055/s-2008-1032159
Return to citation in text: [1] [2] -
Magesh, C. J.; Nagarajan, R.; Karthik, M.; Perumal, P. T. Appl. Catal., A: Gen. 2004, 266, 1–10. doi:10.1016/j.apcata.2004.01.024
Return to citation in text: [1] -
Thirupathi, P.; Kim, S. S. J. Org. Chem. 2010, 75, 5240–5249. doi:10.1021/jo1010137
Return to citation in text: [1] -
Dhiman, S.; Ramasastry, S. S. V. Org. Biomol. Chem. 2013, 11, 8030–8035. doi:10.1039/c3ob41945b
Return to citation in text: [1] -
Pal, R.; Khannobis, S.; Sarkar, T. Chem. J. 2013, 3, 7–12.
Return to citation in text: [1] -
Ramachandiran, K.; Muralidharan, D.; Perumal, P. T. Tetrahedron Lett. 2011, 52, 3579–3583. doi:10.1016/j.tetlet.2011.04.110
Return to citation in text: [1] [2]
| 43. | Bell, R.; Carmeli, S.; Sar, N. J. Nat. Prod. 1994, 57, 1587–1590. doi:10.1021/np50113a022 |
| 44. | Gibbs, T. J. K.; Tomkinson, N. C. O. Org. Biomol. Chem. 2005, 3, 4043–4045. doi:10.1039/b513394g |
| 45. | Guo, X.; Pan, S.; Liu, J.; Li, Z. J. Org. Chem. 2009, 74, 8848–8851. doi:10.1021/jo902093p |
| 46. | Niu, T.; Huang, L.; Wu, T.; Zhang, Y. Org. Biomol. Chem. 2011, 9, 273–277. doi:10.1039/c0ob00709a |
| 47. | Tayebee, R.; Nehzat, F.; Rezaei-Seresht, E.; Mohammadi, F. Z.; Rafiee, E. J. Mol. Catal. A: Chem. 2011, 351, 154–164. doi:10.1016/j.molcata.2011.09.029 |
| 48. | Abe, T.; Nakamura, S.; Yanada, R.; Choshi, T.; Hibino, S.; Ishikura, M. Org. Lett. 2013, 15, 3622–3625. doi:10.1021/ol401486s |
| 49. | Huo, C.; Kang, L.; Xu, X.; Jia, X.; Wang, X.; Xie, H.; Yuan, Y. Tetrahedron Lett. 2014, 55, 954–958. doi:10.1016/j.tetlet.2013.12.055 |
| 50. | CCDC-1007024 (6ad) and CCDC-1007025 (6al) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 56. | Xu, H.-Y.; Zi, Y.; Xu, X.-P.; Wang, S.-Y.; Ji, S.-J. Tetrahedron 2013, 69, 1600–1605. doi:10.1016/j.tet.2012.11.098 |
| 57. | Ramesh, C.; Banerjee, J.; Pal, R.; Das, B. Adv. Synth. Catal. 2003, 345, 557–559. doi:10.1002/adsc.200303022 |
| 54. | Rosenfeld, D. C.; Shekhar, S.; Takemiya, A.; Utsunomiya, M.; Hartwig, J. F. Org. Lett. 2006, 8, 4179–4182. doi:10.1021/ol061174+ |
| 55. | Peng, Y.-Y.; Zhang, Q.-L.; Yuan, J.-J.; Cheng, J.-P. Chin. J. Chem. 2008, 26, 2228–2232. doi:10.1002/cjoc.200890396 |
| 54. | Rosenfeld, D. C.; Shekhar, S.; Takemiya, A.; Utsunomiya, M.; Hartwig, J. F. Org. Lett. 2006, 8, 4179–4182. doi:10.1021/ol061174+ |
| 26. | Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Chem. Rev. 2010, 110, 2250–2293. doi:10.1021/cr900195a |
| 51. | Cook, A. H.; Majer, J. R. J. Chem. Soc. 1944, 486–487. doi:10.1039/JR9440000486 |
| 52. | Cook, A. H.; Majer, J. R. J. Chem. Soc. 1944, 488–489. doi:10.1039/JR9440000488 |
| 53. | Burr, G. O.; Gortner, R. A. J. Am. Chem. Soc. 1924, 46, 1224–1246. doi:10.1021/ja01670a015 |
| 58. | An, L.-T.; Ding, F.-Q.; Zhou, J.-P.; Lu, X.-H.; Zhang, L.-L. Chin. J. Chem. 2007, 25, 822–827. doi:10.1002/cjoc.200790151 |
| 35. | Ji, S.-J.; Zhou, M.-F.; Gu, D.-G.; Jiang, Z.-Q.; Loh, T.-P. Eur. J. Org. Chem. 2004, 1584–1587. doi:10.1002/ejoc.200300719 |
| 59. | Khalafi-Nezhad, A.; Parhami, A.; Zare, A.; Moosavi Zare, A. R.; Hasaninejad, A.; Panahi, F. Synthesis 2008, 617–621. doi:10.1055/s-2008-1032159 |
| 64. | Ramachandiran, K.; Muralidharan, D.; Perumal, P. T. Tetrahedron Lett. 2011, 52, 3579–3583. doi:10.1016/j.tetlet.2011.04.110 |
| 57. | Ramesh, C.; Banerjee, J.; Pal, R.; Das, B. Adv. Synth. Catal. 2003, 345, 557–559. doi:10.1002/adsc.200303022 |
| 62. | Dhiman, S.; Ramasastry, S. S. V. Org. Biomol. Chem. 2013, 11, 8030–8035. doi:10.1039/c3ob41945b |
| 61. | Thirupathi, P.; Kim, S. S. J. Org. Chem. 2010, 75, 5240–5249. doi:10.1021/jo1010137 |
| 58. | An, L.-T.; Ding, F.-Q.; Zhou, J.-P.; Lu, X.-H.; Zhang, L.-L. Chin. J. Chem. 2007, 25, 822–827. doi:10.1002/cjoc.200790151 |
| 23. | Veluri, R.; Oka, I.; Wagner-Döbler, I.; Laatsch, H. J. Nat. Prod. 2003, 66, 1520–1523. doi:10.1021/np030288g |
| 60. | Magesh, C. J.; Nagarajan, R.; Karthik, M.; Perumal, P. T. Appl. Catal., A: Gen. 2004, 266, 1–10. doi:10.1016/j.apcata.2004.01.024 |
| 57. | Ramesh, C.; Banerjee, J.; Pal, R.; Das, B. Adv. Synth. Catal. 2003, 345, 557–559. doi:10.1002/adsc.200303022 |
| 59. | Khalafi-Nezhad, A.; Parhami, A.; Zare, A.; Moosavi Zare, A. R.; Hasaninejad, A.; Panahi, F. Synthesis 2008, 617–621. doi:10.1055/s-2008-1032159 |
| 64. | Ramachandiran, K.; Muralidharan, D.; Perumal, P. T. Tetrahedron Lett. 2011, 52, 3579–3583. doi:10.1016/j.tetlet.2011.04.110 |
| 1. | Ruiz-Sanchis, P.; Savina, S. A.; Albericio, F.; Álvarez, M. Chem. – Eur. J. 2011, 17, 1388–1408. doi:10.1002/chem.201001451 |
| 2. | Gribble, G. W. Top. Heterocycl. Chem. 2010, 26, 1–480. doi:10.1007/978-3-642-15733-2 |
| 3. |
Vicente, R. Org. Biomol. Chem. 2011, 9, 6469–6480. doi:10.1039/C1OB05750B
And references therein cited. |
| 15. | Pindur, U.; Lemster, T. Curr. Med. Chem. 2001, 8, 1681–1698. doi:10.2174/0929867013371941 |
| 16. | Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489–4497. doi:10.1021/cr900211p |
| 29. | Kantam, M. L.; Aziz, K.; Likhar, P. R. Catal. Lett. 2004, 98, 117–121. doi:10.1007/s10562-004-7925-5 |
| 12. | Lounasmaa, M.; Tolvanen, A. Nat. Prod. Rep. 2000, 17, 175–191. doi:10.1039/a809402k |
| 13. | Hibino, S.; Choshi, T. Nat. Prod. Rep. 2001, 18, 66–87. doi:10.1039/b004055j |
| 14. | Wu, Y.-J. Top. Heterocycl. Chem. 2010, 26, 1–29. doi:10.1007/7081_2010_37 |
| 30. | Yu, L.; Chen, D.; Li, J.; Wang, P. G. J. Org. Chem. 1997, 62, 3575–3581. doi:10.1021/jo961933+ |
| 6. | Bandini, M.; Melloni, A.; Tommasi, S.; Umani-Ronchi, A. Synlett 2005, 1199–1222. doi:10.1055/s-2005-865210 |
| 7. | Marqués-López, E.; Diez-Martinez, A.; Merino, P.; Herrera, R. P. Curr. Org. Chem. 2009, 13, 1585–1609. doi:10.2174/138527209789578126 |
| 8. | Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608–9644. doi:10.1002/anie.200901843 |
| 9. | Terrasson, V.; de Figueiredo, R. M.; Campagne, J. M. Eur. J. Org. Chem. 2010, 2635–2655. doi:10.1002/ejoc.200901492 |
| 10. | Zeng, M.; You, S.-L. Synlett 2010, 1289–1301. doi:10.1055/s-0029-1219929 |
| 11. | Chauhan, P.; Chimni, S. S. RSC Adv. 2012, 2, 6117–6134. doi:10.1039/c2ra20544k |
| 27. | Ji, S.-J.; Zhou, M.-F.; Gu, D.-G.; Wang, S.-Y.; Loh, T.-P. Synlett 2003, 2077–2079. doi:10.1055/s-2003-41464 |
| 4. | Joule, J. A. Indole and its Derivatives. In Science of Synthesis; Thomas, E. J., Ed.; Houben-Weyl Methods of Molecular Transformations, Vol. 10; Thieme: Stuttgart, 2000; chapter 10.13. |
| 5. | Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875–2911. doi:10.1021/cr0505270 |
| 28. | Yadav, J. S.; Reddy, B. V. S.; Murthy, C. V. S. R.; Kumar, G. M.; Madan, C. Synthesis 2001, 783–787. doi:10.1055/s-2001-12777 |
| 22. | Lee, C.-H.; Yao, C.-F.; Huang, S.-M.; Ko, S.; Tan, Y.-H.; Lee-Chen, G.-J.; Wang, Y.-C. Cancer 2008, 113, 815–825. doi:10.1002/cncr.23619 |
| 24. | Fischer, E. Chem. Ber. 1886, 19, 2988–2991. doi:10.1002/cber.188601902296 |
| 25. | Fischer, E. Justus Liebigs Ann. Chem. 1887, 242, 372–383. doi:10.1002/jlac.18872420311 |
| 21. | Maciejewska, D.; Niemyjska, M.; Wolska, I.; Włostowski, M.; Rasztawicka, M. Z. Naturforsch. 2004, 59b, 1137–1142. |
| 26. | Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Chem. Rev. 2010, 110, 2250–2293. doi:10.1021/cr900195a |
| 18. | Bell, M. C.; Crowley-Nowick, P.; Bradlow, H. L.; Sepkovic, D. W.; Schmidt-Grimminger, D.; Howell, P.; Mayeaux, E. J.; Tucker, A.; Turbat-Herrera, E. A.; Mathis, J. M. Gynecol. Oncol. 2000, 78, 123–129. doi:10.1006/gyno.2000.5847 |
| 19. | Hong, C.; Firestone, G. L.; Bjeldanes, L. F. Biochem. Pharmacol. 2002, 63, 1085–1097. doi:10.1016/S0006-2952(02)00856-0 |
| 20. | Le, H. T.; Schaldach, C. M.; Firestone, G. L.; Bjeldanes, L. F. J. Biol. Chem. 2003, 278, 21136–21145. doi:10.1074/jbc.M300588200 |
| 17. | Safe, S.; Papineni, S.; Chintharlapalli, S. Cancer Lett. 2008, 269, 326–338. doi:10.1016/j.canlet.2008.04.021 |
| 23. | Veluri, R.; Oka, I.; Wagner-Döbler, I.; Laatsch, H. J. Nat. Prod. 2003, 66, 1520–1523. doi:10.1021/np030288g |
| 33. | Mi, X.; Luo, S.; He, J.; Cheng, J.-P. Tetrahedron Lett. 2004, 45, 4567–4570. doi:10.1016/j.tetlet.2004.04.039 |
| 31. | Soueidan, M.; Collin, J.; Gil, R. Tetrahedron Lett. 2006, 47, 5467–5470. doi:10.1016/j.tetlet.2006.05.169 |
| 32. | Srinivasa, A.; Varma, P. P.; Hulikal, V.; Mahadevan, K. M. Monatsh. Chem. 2008, 139, 111–115. doi:10.1007/s00706-007-0697-z |
| 41. | Hashmi, A. S. K. In Silver in Organic Chemistry; Harmata, M., Ed.; Wiley: Hoboken, 2010; pp 357–379. doi:10.1002/9780470597521.ch12 |
| 42. | Weibel, J.-M.; Blanc, A.; Pale, P. Chem. Rev. 2008, 108, 3149–3173. doi:10.1021/cr078365q |
| 26. | Shiri, M.; Zolfigol, M. A.; Kruger, H. G.; Tanbakouchian, Z. Chem. Rev. 2010, 110, 2250–2293. doi:10.1021/cr900195a |
| 38. | Silveira, C. C.; Mendes, S. R.; Líbero, F. M.; Lenardão, E. J.; Perin, G. Tetrahedron Lett. 2009, 50, 6060–6063. doi:10.1016/j.tetlet.2009.08.062 |
| 39. | Buscemi, G.; Biffis, A.; Tubaro, C.; Basato, M.; Graiff, C.; Tiripicchio, A. Appl. Organomet. Chem. 2010, 24, 285–290. doi:10.1002/aoc.1595 |
| 40. | Shaikh, A. C.; Chen, C. J. Chin. Chem. Soc. 2011, 58, 899–905. doi:10.1002/jccs.201190142 |
| 36. | Shirini, F.; Yahyazadeh, A.; Abedini, M.; Langroodi, D. I. Bull. Korean Chem. Soc. 2010, 31, 1715–1718. doi:10.5012/bkcs.2010.31.6.1715 |
| 37. | Khodaeia, M. M.; Ghanbary, P.; Mohammadpoor-Baltork, I.; Memarian, H. R.; Khosropour, A. R.; Nikoofar, K. J. Heterocycl. Chem. 2008, 45, 377–381. doi:10.1002/jhet.5570450213 |
| 34. | Chen, D.; Yu, L.; Wang, P. G. Tetrahedron Lett. 1996, 37, 4467–4470. doi:10.1016/0040-4039(96)00958-6 |
| 35. | Ji, S.-J.; Zhou, M.-F.; Gu, D.-G.; Jiang, Z.-Q.; Loh, T.-P. Eur. J. Org. Chem. 2004, 1584–1587. doi:10.1002/ejoc.200300719 |
© 2014 Beltrá et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)