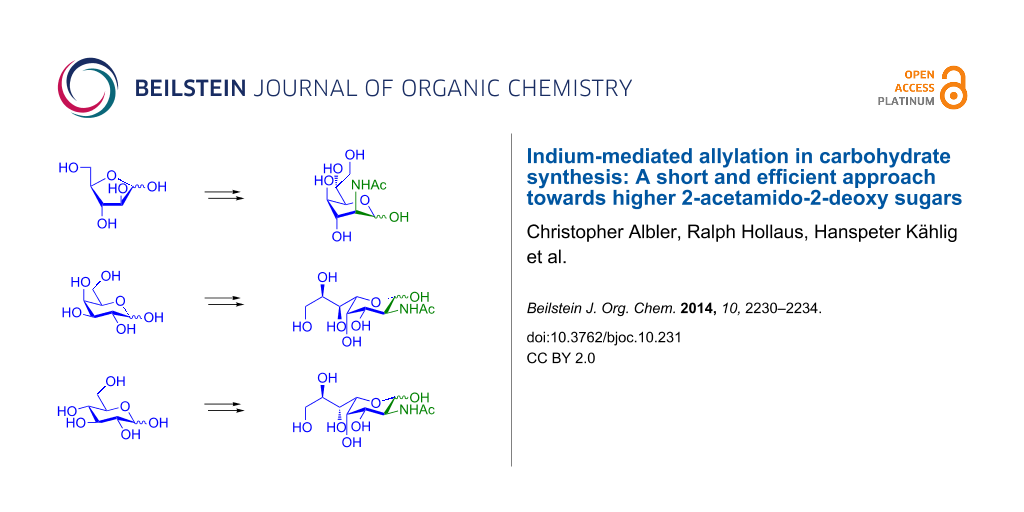
Abstract
Higher aminosugars are interesting targets in carbohydrate synthesis since these compounds play important roles in biological systems. However, their availability from natural sources is limited. Thus, in order to investigate their biological function, the development of facile and adaptable routes to this class of compounds is of fundamental importance. Our synthetic route towards these target molecules makes use of readily accessible pentoses and hexoses, which are subjected to indium-mediated two-carbon chain elongation. Subsequent ozonolysis and treatment with base yields α,β-unsaturated aldehydes, which are stereoselectively epoxidized using Jørgenson’s protocol. After Wittig chain elongation the obtained allylic epoxides were regio- and stereoselectively opened with trimethylsilyl azide under palladium catalysis. Finally, a suitable deprotection protocol, starting with acidic acetate cleavage and ozonolysis was established. Peracetylation of the products simplifies purification and subsequent azide reduction followed by final deacetylation using methanolic sodium methoxide furnishes the title compounds.

Graphical Abstract
Introduction
The indium-mediated allylation of carbonyl compounds has proven to be a valuable tool for carbon chain elongation [1-3] in order to access rare, biologically active carbohydrates [4-8]. Herein we report the extension of this method towards the field of higher aminosugars by additionally applying a stereoselective epoxidation–azide opening strategy. The resulting compounds, aminoheptoses and octoses, have been scarcely investigated yet. However, they comprise interesting synthetic targets. Aminoheptoses function as constituents of the cell wall lipopolysaccharides of certain bacteria [9] whereas their 99mTc complexes find practical applications in medicinal chemistry as tracers for tumor detection, myocardial ischemia or infarct diagnostics [10]. The group of Perez et al has taken a particular interest in the chemistry of 2-aminoheptoses [11-14] which they prepared via an amino-nitrile synthesis [15]. This method, first described by Kuhn and Kirschenlohr [16] for the synthesis of aminohexoses, proved to be straightforward, albeit the reproducibility and yield usually suffered from the formation of multiple side products [17]. Another interesting approach for the synthesis of higher aminosugars, published by Kunz and Deloisy, consists of an aza-Cope rearrangement of N-galactosyl-N-homoallylamines [18]. Aminooctoses, on the other hand, are present in the aminoglycoside antibiotic apramycin [19], in the form of an aminooctodiose derivative. However, only few syntheses of this dipyranoid aminosugar [20,21] were reported so far. Thus, we were interested in developing a general route towards the synthesis of these higher aminosugars. Since the preparation of new potent aminoglycoside antibiotics remains an important topic in medicinal chemistry [22], our precursors used should be flexible in terms of stereochemical variety, thus potentially allowing for an evaluation of structure–activity relationships.
Results and Discussion
We started our reaction sequence with an indium-mediated allylation of unprotected carbohydrates using D-arabinose (1a), D-galactose (1b) and D-glucose (1c) as starting materials. The Barbier-type chain elongation reaction furnished olefins 2a and 2b after acetylation in quantitative yield (Scheme 1). In the gluco-case a yield of 70% for 2c was obtained owing to incomplete consumption of the starting material 1c. Ozonolysis of the olefins 2 was performed using thiourea as reducing agent. Subsequently the generated 2-deoxyaldoses were treated with triethylamine (TEA) to yield α,β-unsaturated aldehydes 3a–c quantitatively.
![[1860-5397-10-231-i1]](/bjoc/content/inline/1860-5397-10-231-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Functionalization of carbohydrates; reagents and conditions: (a) In, allyl bromide, EtOH/H2O, ultrasound, 2.5–7 h, then Ac2O/pyridine, DMAP, rt, 16 h, quant.; (b) O3, CH2Cl2, −78 °C then thiourea, rt, 16 h, then TEA, rt, 30–50 min, quant.; (c) H2O2, cat., CH2Cl2, −20 °C, 16 h, then Ph3P(CHCO2Me), CH2Cl2, rt, 1 h, 72–75%, 2 steps; (d) TMSN3, Pd(PPh3)4, THF, rt, 1 h, 76–85%.
Scheme 1: Functionalization of carbohydrates; reagents and conditions: (a) In, allyl bromide, EtOH/H2O, ultra...
The aldehydes 3a–c were stereoselectively epoxidized by applying the conditions developed by Jørgenson et al [23-25]. The required amine catalyst was synthesized starting from L-proline following a literature procedure [26]. In general the desired epoxyaldehydes were obtained with high diastereoselectivity at low temperatures (Table 1). The stereochemistry of compounds 3a–c is in accordance with the proposed reaction mechanism [24] and was proven by applying various NMR methods on the final products (vide infra), which adopt rigid pyranoid conformations.
Table 1: Epoxidation of aldehydes 3a–c.
| entry | aldehyde | dra | yieldb [%] |
|---|---|---|---|
| 1 | 3a | 9:1 | 64 |
| 2 | 3b | 99:1 | 77 |
| 3 | 3c | 9:1 | 61 |
aDetermined either by comparison of integrals of representative 1H NMR signals of crude reaction products or after conventional column chromatographic separation of diastereomers; bisolated yields of corresponding 2,3-epoxyaldehydes.
Unfortunately, we were not able to directly achieve the opening of the obtained 2,3-epoxyaldehydes with azide nucleophiles. A variety of different Lewis acids, solvent systems and azide sources were screened but all attempts led to the decomposition of these labile compounds. Conversion of the carbonyl moiety to an acetal group followed by treatment with azide nucleophiles also failed to yield any desired products. Therefore we decided to mask the aldehyde as an olefin. A Wittig reaction with methyl (triphenylphosphoranylidene)acetate (Ph3P(CHCO2Me)) generated allylic epoxides 4a–c, which in turn permitted the application of reliable palladium chemistry for the epoxide opening [27-29]. Thus, compounds 4a–c were regio- and stereoselectively opened with trimethylsilyl azide and Pd(PPh3)4 as a catalyst [30], furnishing syn-azido alcohols 5a–c.
In the case of allylic azide 5a we observed an acetate migration which resulted in a complex mixture of products bearing a free hydroxy moiety (mixture of free C5-OH/C6-OH/C7-OH = 2/1/1; Scheme 1). We reasoned that this migration was limited to the inherent syn relationship between C5-OH and C6-OAc in compound 5a, since this behavior was not observed with compounds 5b and 5c which feature an anti alignment of the hydroxy groups. Next, we investigated the reduction of the azide moiety which should be followed by cleavage of the acetate protecting groups and ozonolysis in order to generate the target compounds. However, we found that compounds 5a–c easily underwent 1,4-additions in the presence of nucleophiles or were decomposed under basic conditions. In this respect, DL-dithiothreitol/diisopropylamine (DTT/DIPA), DIBALH and NaBH4/MeOH failed to furnish the desired reduction products. Treatment with Pd/H2, PPh3, P(OMe)3 or SnCl2 on the other hand resulted in no or only very low conversion. Thus, we decided to change the reaction sequence. Consequently, we performed the acetate cleavage as the first step, since direct ozonolysis of compounds 5a and 5c again resulted in decomposition. Deacetylation in a methanolic HCl solution proved to be most efficient and the subsequent ozonolysis generated sugar azides 6a–c (Scheme 2).
![[1860-5397-10-231-i2]](/bjoc/content/inline/1860-5397-10-231-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Deprotection sequence; reagents and conditions: (a): HCl/MeOH, rt, 16–24 h, then MeOH, CH2Cl2, O3, −78 °C then PPh3, rt, 16 h, 71–82%, 2 steps; (b): Ac2O/pyridine, DMAP, rt, 16 h then CH3CN, DTT, DIPA, rt, 2 h, then Ac2O/pyridine, DMAP, rt, 16 h, 59–66%, 3 steps; (c): NaOMe/MeOH, rt, 2–3 h, quant.
Scheme 2: Deprotection sequence; reagents and conditions: (a): HCl/MeOH, rt, 16–24 h, then MeOH, CH2Cl2, O3, ...
It turned out to be essential to perform the acidic acetate cleavage of compounds 5a–c under strictly dry conditions, since even small amounts of water promoted an intramolecular Michael addition, leading to the formation of highly substituted tetrahydrofuran derivatives of the C-glycoside type [31].
Next, we investigated the azide reduction of compounds 6a–c under different reaction conditions. Unfortunately DTT/DIPA, thioacetic acid, tributylphosphine/H2O and H2/Pd followed by N-acetylation with N-acetoxyphthalimide did not afford products 8a–c in acceptable yields and/or purity. Since compounds 8a–c could not be purified by conventional silica gel chromatography we decided to perform an additional acetylation step in order to avoid reversed-phase HPLC and to maintain our procedure as simple as possible. Hence, compounds 6a–c were acetylated under standard conditions, subsequently reduced with DTT/DIPA and N-acetylated to yield compounds 7a–c which could be easily purified. Final treatment with sodium methoxide in dry methanol then furnished the neat title compounds 8a–c.
The configurations of compounds 6, 7 and 8 were determined by applying various NMR methods. Since the stereochemistry at C-4 is given by the starting material, configurations of C-2 and C-3 can be easily deduced from the magnitude of the respective 3JH,H coupling constants which reflect the torsion angles between these groups (Table 2).
Table 2: Characteristic coupling constants of compounds 7a–c.
| Compound | 3J1,2 [Hz] | 3J2,3 [Hz] | 3J3,4 [Hz] | 3J4,5 [Hz] |
|---|---|---|---|---|
| 7a-αa | 1.8 | 3.1 | 3.1 | 1.9 |
| 7a-βa | 2.1 | 2.9 | 2.9 | 1.8 |
| 7b-α | 3.7 | 11.5 | 3.3 | 0.9 |
| 7b-β | 9.0 | 11.3 | 3.5 | 1.1 |
| 7c-α | 3.7 | 11.6 | 3.3 | 1.0 |
| 7c-β | 8.8 | 11.1 | 3.3 | 0.5 |
a 4C1-pyranoid form.
Therefore, the anticipated configuration of the final products could be verified. Since compounds 6a and 8a were found to exist as complex mixtures of conformers, only anomeric signals were assigned. This finding is in agreement with the fact that ido-configurated pyranose derivatives are known to adopt multiple conformations [32-34]. We were able to isolate and fully assign four distinct forms of the acetylated monosaccharide derivative 7a (β-furanoid, 4C1 α/β-pyranoid and 1C4 α-pyranoid). Although 7a is a known compound [15], the published NMR data are scarce. Herein, we provide the complete NMR data sets of 2-acetamido-1,3,4,6,7-penta-O-acetyl-2-deoxy-D-glycero-D-ido-heptose (7a). In the cases of compounds 6b, 6c, 7b, 7c and 8b only α,β-pyranoid forms were detected (see copies of NMR spectra provided in Supporting Information File 1). However, NMR spectra of 8c showed minor amounts of a compound which we considered to be its corresponding furanoid form. Unfortunately, we were not able to confirm this hypothesis owing to the very low concentration of this compound.
Conclusion
In summary, we developed a simple, highly versatile route for the synthesis of rare, 2-amino-functionalized heptoses and octoses. The indium-mediated allylation strategy again revealed to be a useful tool for the preparation of two-carbon chain elongated carbohydrates. Two new stereocenters were formed with high diastereoselectivity in the course of the synthesis owing to the use of a chiral L-proline derived epoxidation catalyst. The introduction of nitrogen was achieved via a Tsuji–Trost-like azide opening of allylic epoxides. Although global deprotection proved to be cumbersome, we were able to develop a versatile reaction sequence to overcome this problem. The desired higher aminosugars were obtained in an overall yield of 21–29% over 7 steps. Additionally, these compounds should now be available in all stereochemical combinations by means of varying the starting materials, the epoxidation catalysts, or the mode of azide opening, respectively [35]. Furthermore, biologically active octodioses are now accessible from compounds 7b and 7c, which is subject of ongoing research.
Supporting Information
| Supporting Information File 1: Experimental section, spectral data and copies of 1H and 13C NMR spectra of compounds 2–8. | ||
| Format: PDF | Size: 3.3 MB | Download |
References
-
Li, C.-J.; Chan, T.-H. Tetrahedron 1999, 55, 11149–11176. doi:10.1016/S0040-4020(99)00641-9
Return to citation in text: [1] -
Araki, S.; Hirashita, T. Indium in Organic Synthesis. In Main Group Metals in Organic Synthesis; Yamamoto, H.; Oshima, K., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Augé, J.; Lubin-Germain, N.; Uziel, J. Synthesis 2007, 1739–1764. doi:10.1055/s-2007-983703
Return to citation in text: [1] -
Chan, T.-H.; Li, C.-J. J. Chem. Soc., Chem. Commun. 1992, 747–748. doi:10.1039/c39920000747
Return to citation in text: [1] -
Gao, J.; Härter, R.; Gordon, D. M.; Whitesides, G. M. J. Org. Chem. 1994, 59, 3714–3715. doi:10.1021/jo00092a040
Return to citation in text: [1] -
Prenner, R. H.; Binder, W. H.; Schmid, W. Liebigs Ann. Chem. 1994, 73–78. doi:10.1002/jlac.199419940113
Return to citation in text: [1] -
Schmölzer, C.; Fischer, M.; Schmid, W. Eur. J. Org. Chem. 2010, 4886–4892. doi:10.1002/ejoc.201000623
Return to citation in text: [1] -
Lee, Y. J.; Kubota, A.; Ishiwata, A.; Ito, Y. Tetrahedron Lett. 2011, 52, 418–421. doi:10.1016/j.tetlet.2010.11.078
Return to citation in text: [1] -
Weise, G.; Drews, G.; Jann, B.; Jann, K. Arch. Microbiol. 1970, 71, 89–98.
Return to citation in text: [1] -
Steinmetz, H. J.; Schwochau, K. Technetium complexes, processes for the preparation thereof and application kits for the formation of technetium complexes. Ger. Patent 4,128,181, Feb 29, 1996.
Return to citation in text: [1] -
Gonzáles, F. G.; Guillen, M. G.; Pérez, J. A. G.; Galán, E. R. Carbohydr. Res. 1980, 78, 17–23. doi:10.1016/S0008-6215(00)83656-3
Return to citation in text: [1] -
Pérez, J. A. G.; Albarrán, J. C. P.; Galán, E. R. Carbohydr. Res. 1983, 114, 158–163. doi:10.1016/0008-6215(83)88182-8
Return to citation in text: [1] -
Perez, J. A. G.; Caballero, R. B.; Ventula, A. C. Carbohydr. Res. 1985, 143, 129–141. doi:10.1016/S0008-6215(00)90702-X
Return to citation in text: [1] -
Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J. L.; Palacios, J. C.; Valencia, C. Tetrahedron 1993, 49, 2676–2690. doi:10.1016/S0040-4020(01)86345-6
Return to citation in text: [1] -
Perez, J. A. G.; Corraliza, R. M. P.; Galan, E. R.; Guillen, M. G. An. Quim. (1968–1979) 1979, 75, 387–391.
Return to citation in text: [1] [2] -
Kuhn, R.; Kirschenlohr, W. Angew. Chem. 1955, 67, 786. doi:10.1002/ange.19550672405
Return to citation in text: [1] -
Albarran, J. C. P.; Galan, E. R.; Perez, J. A. G. Carbohydr. Res. 1985, 143, 117–128. doi:10.1016/S0008-6215(00)90701-8
Return to citation in text: [1] -
Deloisy, S.; Kunz, H. Tetrahedron Lett. 1998, 39, 791–794. doi:10.1016/S0040-4039(97)10720-1
Return to citation in text: [1] -
Matt, T.; Ng, C. L.; Lang, K.; Sha, S.-H.; Akbergenov, R.; Shcherbakov, D.; Meyer, M.; Duscha, S.; Xie, J.; Dubbaka, S. R.; Perez-Fernandez, D.; Vasella, A.; Ramakrishnan, V.; Schacht, J.; Böttger, E. C. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 10984–10989. doi:10.1073/pnas.1204073109
Return to citation in text: [1] -
Tatsuta, K.; Akimoto, K.; Takahashi, H.; Hamatsu, T.; Annaka, M.; Kinoshita, M. Tetrahedron Lett. 1983, 24, 4867–4870. doi:10.1016/S0040-4039(00)94028-0
Return to citation in text: [1] -
Martin, O. R.; Szarek, W. A. Carbohydr. Res. 1984, 130, 195–219. doi:10.1016/0008-6215(84)85280-5
Return to citation in text: [1] -
Fair, R. J.; Hensler, M. E.; Thienphrapa, W.; Dam, Q. N.; Nizet, V.; Tor, Y. ChemMedChem 2012, 7, 1237–1244. doi:10.1002/cmdc.201200150
Return to citation in text: [1] -
Marigo, M.; Franzén, J.; Poulsen, T. B.; Zhuang, W.; Jørgensen, K. A. J. Am. Chem. Soc. 2005, 127, 6964–6965. doi:10.1021/ja051808s
Return to citation in text: [1] -
Zhao, G.-L.; Ibrahem, I.; Sundén, H.; Córdova, A. Adv. Synth. Catal. 2007, 349, 1210–1224. doi:10.1002/adsc.200600529
Return to citation in text: [1] [2] -
Jensen, K. L.; Dickmeiss, G.; Jiang, H.; Albrecht, L.; Jørgensen, K. A. Acc. Chem. Res. 2012, 45, 248–264. doi:10.1021/ar200149w
Return to citation in text: [1] -
Kanth, J. V. B.; Periasamy, M. Tetrahedron 1993, 49, 5127–5132. doi:10.1016/S0040-4020(01)81877-9
Return to citation in text: [1] -
Trost, B. M.; Bunt, R. C.; Lemoine, R. C.; Calkins, T. L. J. Am. Chem. Soc. 2000, 122, 5968–5976. doi:10.1021/ja000547d
Return to citation in text: [1] -
Trost, B. M.; Jiang, C.; Hammer, K. Synthesis 2005, 3335–3345. doi:10.1055/s-2005-918443
Return to citation in text: [1] -
Trost, B. M.; Horne, D. B.; Woltering, M. J. Chem. – Eur. J. 2006, 12, 6607–6620. doi:10.1002/chem.200600202
Return to citation in text: [1] -
Miyashita, M.; Mizutani, T.; Tadano, G.; Iwata, Y.; Miyazawa, M.; Tanino, K. Angew. Chem., Int. Ed. 2005, 44, 5094–5097. doi:10.1002/anie.200500838
Return to citation in text: [1] -
The structure of prepared C-glycosides was determined by NMR, mass and IR spectroscopy although the final proof for the stereochemistry of these compounds is a matter of ongoing research.
Return to citation in text: [1] -
Bhacca, N. S.; Horton, D.; Paulsen, H. J. Org. Chem. 1968, 33, 2484–2487. doi:10.1021/jo01270a068
Return to citation in text: [1] -
Horita, D. A.; Hajduk, P. J.; Lerner, L. E. Glycoconjugate J. 1997, 14, 691–696. doi:10.1023/A:1018561215401
Return to citation in text: [1] -
Kräutler, V.; Müller, M.; Hünenberger, P. H. Carbohydr. Res. 2007, 342, 2097–2124. doi:10.1016/j.carres.2007.05.011
Return to citation in text: [1] -
Righi, G.; Manni, L. S.; Bovicelli, P.; Pelagalli, R. Tetrahedron Lett. 2011, 52, 3895–3896. doi:10.1016/j.tetlet.2011.05.085
Return to citation in text: [1]
| 32. | Bhacca, N. S.; Horton, D.; Paulsen, H. J. Org. Chem. 1968, 33, 2484–2487. doi:10.1021/jo01270a068 |
| 33. | Horita, D. A.; Hajduk, P. J.; Lerner, L. E. Glycoconjugate J. 1997, 14, 691–696. doi:10.1023/A:1018561215401 |
| 34. | Kräutler, V.; Müller, M.; Hünenberger, P. H. Carbohydr. Res. 2007, 342, 2097–2124. doi:10.1016/j.carres.2007.05.011 |
| 30. | Miyashita, M.; Mizutani, T.; Tadano, G.; Iwata, Y.; Miyazawa, M.; Tanino, K. Angew. Chem., Int. Ed. 2005, 44, 5094–5097. doi:10.1002/anie.200500838 |
| 31. | The structure of prepared C-glycosides was determined by NMR, mass and IR spectroscopy although the final proof for the stereochemistry of these compounds is a matter of ongoing research. |
| 1. | Li, C.-J.; Chan, T.-H. Tetrahedron 1999, 55, 11149–11176. doi:10.1016/S0040-4020(99)00641-9 |
| 2. | Araki, S.; Hirashita, T. Indium in Organic Synthesis. In Main Group Metals in Organic Synthesis; Yamamoto, H.; Oshima, K., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. |
| 3. | Augé, J.; Lubin-Germain, N.; Uziel, J. Synthesis 2007, 1739–1764. doi:10.1055/s-2007-983703 |
| 11. | Gonzáles, F. G.; Guillen, M. G.; Pérez, J. A. G.; Galán, E. R. Carbohydr. Res. 1980, 78, 17–23. doi:10.1016/S0008-6215(00)83656-3 |
| 12. | Pérez, J. A. G.; Albarrán, J. C. P.; Galán, E. R. Carbohydr. Res. 1983, 114, 158–163. doi:10.1016/0008-6215(83)88182-8 |
| 13. | Perez, J. A. G.; Caballero, R. B.; Ventula, A. C. Carbohydr. Res. 1985, 143, 129–141. doi:10.1016/S0008-6215(00)90702-X |
| 14. | Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J. L.; Palacios, J. C.; Valencia, C. Tetrahedron 1993, 49, 2676–2690. doi:10.1016/S0040-4020(01)86345-6 |
| 24. | Zhao, G.-L.; Ibrahem, I.; Sundén, H.; Córdova, A. Adv. Synth. Catal. 2007, 349, 1210–1224. doi:10.1002/adsc.200600529 |
| 10. | Steinmetz, H. J.; Schwochau, K. Technetium complexes, processes for the preparation thereof and application kits for the formation of technetium complexes. Ger. Patent 4,128,181, Feb 29, 1996. |
| 27. | Trost, B. M.; Bunt, R. C.; Lemoine, R. C.; Calkins, T. L. J. Am. Chem. Soc. 2000, 122, 5968–5976. doi:10.1021/ja000547d |
| 28. | Trost, B. M.; Jiang, C.; Hammer, K. Synthesis 2005, 3335–3345. doi:10.1055/s-2005-918443 |
| 29. | Trost, B. M.; Horne, D. B.; Woltering, M. J. Chem. – Eur. J. 2006, 12, 6607–6620. doi:10.1002/chem.200600202 |
| 23. | Marigo, M.; Franzén, J.; Poulsen, T. B.; Zhuang, W.; Jørgensen, K. A. J. Am. Chem. Soc. 2005, 127, 6964–6965. doi:10.1021/ja051808s |
| 24. | Zhao, G.-L.; Ibrahem, I.; Sundén, H.; Córdova, A. Adv. Synth. Catal. 2007, 349, 1210–1224. doi:10.1002/adsc.200600529 |
| 25. | Jensen, K. L.; Dickmeiss, G.; Jiang, H.; Albrecht, L.; Jørgensen, K. A. Acc. Chem. Res. 2012, 45, 248–264. doi:10.1021/ar200149w |
| 4. | Chan, T.-H.; Li, C.-J. J. Chem. Soc., Chem. Commun. 1992, 747–748. doi:10.1039/c39920000747 |
| 5. | Gao, J.; Härter, R.; Gordon, D. M.; Whitesides, G. M. J. Org. Chem. 1994, 59, 3714–3715. doi:10.1021/jo00092a040 |
| 6. | Prenner, R. H.; Binder, W. H.; Schmid, W. Liebigs Ann. Chem. 1994, 73–78. doi:10.1002/jlac.199419940113 |
| 7. | Schmölzer, C.; Fischer, M.; Schmid, W. Eur. J. Org. Chem. 2010, 4886–4892. doi:10.1002/ejoc.201000623 |
| 8. | Lee, Y. J.; Kubota, A.; Ishiwata, A.; Ito, Y. Tetrahedron Lett. 2011, 52, 418–421. doi:10.1016/j.tetlet.2010.11.078 |
| 26. | Kanth, J. V. B.; Periasamy, M. Tetrahedron 1993, 49, 5127–5132. doi:10.1016/S0040-4020(01)81877-9 |
| 18. | Deloisy, S.; Kunz, H. Tetrahedron Lett. 1998, 39, 791–794. doi:10.1016/S0040-4039(97)10720-1 |
| 20. | Tatsuta, K.; Akimoto, K.; Takahashi, H.; Hamatsu, T.; Annaka, M.; Kinoshita, M. Tetrahedron Lett. 1983, 24, 4867–4870. doi:10.1016/S0040-4039(00)94028-0 |
| 21. | Martin, O. R.; Szarek, W. A. Carbohydr. Res. 1984, 130, 195–219. doi:10.1016/0008-6215(84)85280-5 |
| 17. | Albarran, J. C. P.; Galan, E. R.; Perez, J. A. G. Carbohydr. Res. 1985, 143, 117–128. doi:10.1016/S0008-6215(00)90701-8 |
| 22. | Fair, R. J.; Hensler, M. E.; Thienphrapa, W.; Dam, Q. N.; Nizet, V.; Tor, Y. ChemMedChem 2012, 7, 1237–1244. doi:10.1002/cmdc.201200150 |
| 16. | Kuhn, R.; Kirschenlohr, W. Angew. Chem. 1955, 67, 786. doi:10.1002/ange.19550672405 |
| 15. | Perez, J. A. G.; Corraliza, R. M. P.; Galan, E. R.; Guillen, M. G. An. Quim. (1968–1979) 1979, 75, 387–391. |
| 15. | Perez, J. A. G.; Corraliza, R. M. P.; Galan, E. R.; Guillen, M. G. An. Quim. (1968–1979) 1979, 75, 387–391. |
| 19. | Matt, T.; Ng, C. L.; Lang, K.; Sha, S.-H.; Akbergenov, R.; Shcherbakov, D.; Meyer, M.; Duscha, S.; Xie, J.; Dubbaka, S. R.; Perez-Fernandez, D.; Vasella, A.; Ramakrishnan, V.; Schacht, J.; Böttger, E. C. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 10984–10989. doi:10.1073/pnas.1204073109 |
| 35. | Righi, G.; Manni, L. S.; Bovicelli, P.; Pelagalli, R. Tetrahedron Lett. 2011, 52, 3895–3896. doi:10.1016/j.tetlet.2011.05.085 |
© 2014 Albler et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)