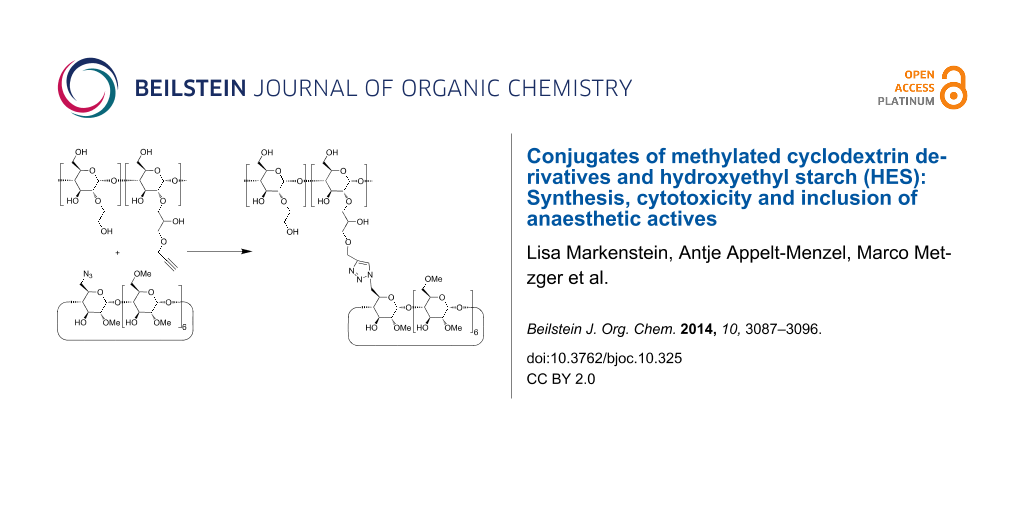
Abstract
The mono-6-deoxy-6-azides of 2,6-di-O-methyl-β-cyclodextrin (DIMEB) and randomly methylated-β-cyclodextrin (RAMEB) were conjugated to propargylated hydroxyethyl starch (HES) by Cu+-catalysed [2 + 3] cycloaddition. The resulting water soluble polymers showed lower critical solution temperatures (LCST) at 52.5 °C (DIMEB-HES) and 84.5 °C (RAMEB-HES), respectively. LCST phase separations could be completely avoided by the introduction of a small amount of carboxylate groups at the HES backbone. The methylated CDs conjugated to the HES backbone exhibited significantly lower cytotoxicities than the corresponding monomeric CD derivatives. Since the binding potentials of these CD conjugates were very high, they are promising candidates for new oral dosage forms of anaesthetic actives.

Graphical Abstract
Introduction
Cyclodextrins (CDs), α(1→4) linked cyclic oligomers of anhydroglucose, are produced nowadays in industrial scale [1]. CDs are able to complex hydrophobic or amphiphilic guest molecules in aqueous phase [2]. β-CD, the seven membered ring forms inclusion compounds with derivatives of benzene, naphthalene, adamantane, and many other moieties of similar sizes [3]. Applications of β-CD as complexing agents are limited because of low aqueous solubilities of β-CD and its complexes. Furthermore, the toxic potential of β-CD is known for a long time. β-CD can cause haemolysis due to extraction of cholesterol from cell walls [4,5]. Also the parenteral application of high doses of β-CD can cause kidney diseases [6,7]. Consequently, pharmaceutical applications of β-CD are restricted to oral dosage forms, such as piroxicam β-CD [8-10].
Toxicity of β-CD can be minimized by derivatisation [11,12]. Both, hydroxypropyl-β-CD (HP-β-CD) and sulfobutyl-β-CD are less toxic than β-CD, but they are less defined due to a statistical substitution pattern [13]. HP-β-CD often shows a reduced binding potential compared to β-CD [2,14]. On the other hand, methylation of β-CD leads to excellent solubilities in water and high binding potentials, but causes even higher toxicity compared to native β-CD. Among the methyl derivatives of β-CD the heptakis-2,6-di-O-methyl derivative, abbreviated as DIMEB [15], showed very high binding potentials [16] and was already discussed by Szejli as a very promising candidate for parenteral drug delivery [17], but it was placed back because of its high toxicity [5,18,19]. Therefore our aim was to conjugate DIMEB for the first time to a polymeric backbone, which should hinder cellular uptake. CD polymers are known to have a much lower toxic potential compared to CD monomers [20].
Native β-CD was already conjugated by esterification [21], reductive amination [22-25], amide coupling [26,27] and [2 + 3] cycloadditions [28] to both biogenic and synthetic polymers. Conjugation of CDs to polysaccharides like chitosane [29], alginate [24] and dextrane [28] is advantageous for the design of drug delivery systems because of the low toxicities and biodegradabilities of those polymers. Among the various coupling reactions, the [2 + 3] cycloaddition of alkynes and azides, the so-called Huisgen reaction [30] and its Cu+-catalyzed version, called click reaction [31,32], is of special interest because this coupling proceeds rapidly and specific in aqueous media and tolerates many functional groups. Mono-6-azido-6-deoxy-β-CD was already coupled by the click reaction to propargylated dextrane by Nielsen et al. [28]. We intended to conjugate the corresponding methylated mono-azido-β-CD derivatives to propargylated hydroxyethyl starch (HES). Advantages of HES are its very high aqueous solubility paired with good biocompatibility and its very low allergenic potential [33]. HES is in use for a long time for many parenteral applications, such as plasma expanders [34]. Its rate of bio-degradation increases with decreasing the degree of substitution (MS) of the hydroxyethyl side groups. Nowadays, HES with a molecular weight of Mw = 130 kDa and MS <0.5 is preferred [35]. In the following, we describe the conjugation of azido-functionalized methylated β-CD derivatives to propargylated HES, the evaluation of the toxicity of these new polymers and first binding studies for the hydrophobic anaesthetic ingredients sevoflurane and midazolam. Sevoflurane, currently applied as an inhalation anaesthetic [36], could be solubilized in water to allow further use in oral or parenteral dosage forms as an analgesic drug. Also the uptake of midazolam could be improved by complexation in CD derivatives [37,38].
Results and Discussion
Cyclodextrin polymers were synthesized by copper-catalyzed [2 + 3] cycloaddition of methylated derivatives of mono-6-azido-6-deoxy-β-CD (β-CD-N3) and propargylated hydroxyethyl starch (HES). Furthermore, a partially oxidized propargylated HES was employed as hydrophilic polymer backbone.
The methylated β-CD-N3 was synthesized in a 3 step procedure starting from β-CD which was first converted via the 6-O-tosylate to β-CD-N3 following the procedures of Hocquelet et al. who also described the permethylated β-CD-N3 [39]. Furthermore, we headed for the regioselective 2,6-dimethylated product. Therefore β-CD-N3 was carefully methylated by dimethyl sulfate and a mixture of Ba(OH)2·8H2O and BaO as the base following the procedure of Szejtli et al. published for the methylation of native β-CD [15] (Scheme 1).
![[1860-5397-10-325-i1]](/bjoc/content/inline/1860-5397-10-325-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of azido functionalized DIMEB 1a. a) Ba(OH)2·8H2O/BaO/Me2SO4
Scheme 1: Synthesis of azido functionalized DIMEB 1a. a) Ba(OH)2·8H2O/BaO/Me2SO4
The product 1a showed a quite narrow distribution of methyl substituents as revealed by the ESIMS showing signals corresponding to β-CD-N3 derivatives with 14 to 18 methyl groups (Figure 1). From the average number of the molecular weight obtained from the ESIMS, Mn = 1383.6 g mol−1 a degree of substitution of methyl groups DSCD(methyl) = 2.2 per glucose unit was derived.
![[1860-5397-10-325-1]](/bjoc/content/figures/1860-5397-10-325-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ESI mass spectrum of azido functionalized DIMEB 1a.
Figure 1: ESI mass spectrum of azido functionalized DIMEB 1a.
This value of DSCD(methyl) was confirmed by the well resolved 1H NMR spectrum (Figure 2). After setting the integral of the H-3 proton to 1 the signals in the range between 4.8 and 5.4 ppm integrated to a value of 12.18 protons. Taking into account the 7 anomeric protons 5.18 OH protons per CD are left, which is equivalent to a DSCD(methyl) = 2.2. The signals at 3.66 ppm and 3.43 ppm were assigned to the methyl groups in O-2 and O-6 position, respectively [40]. Nearly no signal was found corresponding to methyl groups in O-3 position. If the according methylation was instead performed with NaOH as the base it proceeded much faster and with a high chance of over-methylation. Careful control of the reaction time lead to another product 1b with a similar degree of methylation DSCD(methyl) = 2.3, but a much higher structural heterogeneity as demonstrated by the 1H NMR spectrum (Figure 3s, Supporting Information File 2) which was much less resolved than the spectrum of 1a (Figure 2a). Methylations employing NaOH as the base [41] are known to be less regioselective than those using barium hydroxide [41].
![[1860-5397-10-325-2]](/bjoc/content/figures/1860-5397-10-325-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectra of a) 1a, b) 2, and c) 5a.
Figure 2: 1H NMR spectra of a) 1a, b) 2, and c) 5a.
A commercial HES with an average molar mass Mw = 130 kDa and a molar degree of substitution of MS(hydroxyethyl) = 0.4 was functionalized by reaction with propargyl glycidyl ether in water (Scheme 2) in analogy to a procedure published by Nielsen et al. for the synthesis of β-CD-dextran polymers [28]. The degree of substitution of HES with propargyl groups, DSHES(propg), was determined by 1H NMR spectroscopy from the integral of the methylene protons of the propargyl groups H-12 at 4.75 ppm relative to the ones of the anomeric proton H-01 of starch at around 5.5 ppm (Figure 2b). Surprisingly DSHES(propg) increased with decreasing concentration of the base NaOH. Only a moderate DSHES(propg) = 0.4 could be reached in 0.75 M NaOH, while a higher DSHES(propg) = 0.65 was accomplished in 0.1 M NaOH after 2 d reaction time. The decreasing yield was rationalized by the increasing consumption of propargyl glycidyl ether by hydrolysis with increasing OH− concentration. On the other hand, due to the low pKa ≈ 12 of polyglucanes, deprotonation still takes place at very low concentrations of OH− [42,43]. Indeed the modification of HES seems to take place exclusively at the unsubstituted glucose units, because the signal of the anomeric proton H-01 of the unsubstituted glucose unit at 5.4 ppm [44] of HES (see Figure 1s, Supporting Information File 2) had nearly completely vanished in the product spectrum shown in Figure 2b. The HES derivative with DSHES(propg) = 0.65, 2, was selected for the further coupling to azido-CD derivatives. Also a hydroxyethyl starch, where the CH2OH groups had been partially oxidized by TEMPO to carboxylate groups [45,46], was functionalized by reaction with glycidyl propargyl ether leading to a highly water soluble polymer 3 with DSHES(propargyl) = 0.55.
![[1860-5397-10-325-i2]](/bjoc/content/inline/1860-5397-10-325-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of propargylated HES 2.
Scheme 2: Synthesis of propargylated HES 2.
Coupling of the CD azides 1a and 1b to the propargylated HES was performed by Cu+-catalyzed [2 + 3] cycloaddition, the so-called click reaction, leading to the corresponding triazol groups (Scheme 3). The standard protocol, introduced by Sharpless [31], using CuSO4 plus ascorbic acid as the catalyst already gave rise to high coupling yields. The resulting conjugates of HES and methylated CDs were isolated by ultrafiltration in nearly quantitative yields (Table 1). Nearly all propargyl groups at the HES detectable by the 1H NMR signal H-12 at 4.26 pm were converted to triazole groups (1H NMR signal H-14 at 8.03 ppm), shown in Figure 2c. The corresponding IR spectra (Figure 5s, Supporting Information File 2) revealed that the excess of azido-CD had been completely removed by the ultrafiltration step since the band of the azido group at 2102 cm−1 was not detectable anymore in the product. The degree of substitution of HES by CD, DSHES(CD), was determined from the ratio of the 1H NMR signals of H-14 at 8.06 ppm (0.38 protons) and of the anomeric proton H-01 of starch at 5.66 ppm.
![[1860-5397-10-325-i3]](/bjoc/content/inline/1860-5397-10-325-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 5a by [2 + 3] cycloadditon, a) CuSO4, ascorbate, 50 °C, 48 h.
Scheme 3: Synthesis of 5a by [2 + 3] cycloadditon, a) CuSO4, ascorbate, 50 °C, 48 h.
All of the methylated CD-HES conjugates, listed in Table 1, were clearly soluble in water at ambient conditions but most of them precipitated at elevated temperatures. This so-called lower critical solution temperature is typical for alkylated neutral polysaccharides and attributed to increasing hydrophobic interactions with increasing temperatures [47-49]. Only the CD-HES conjugate 6 did not show any precipitation below 100 °C which was attributed to the much higher hydrophilicity of the anionic carboxylate groups at the HES backbone compared to unmodified HES.
Cytotoxicity assays
The effect of the CD polymer 5a on the cell viability was assessed using the ATP-based CellTiter-Glo® assay [50] on the human colon tumor cell line Caco-2. A first series of tests was performed in the relevant concentration range, i.e., 10× lower and up to about 10× above the clinically relevant concentration, namely 0.25 till 25 mg/mL of the polymer 5a in the medium for 2 h and 24 h incubation times, respectively (Figure 3a,b). For comparison, the same viability test was carried out with DIMEB (Figure 3c,d).
![[1860-5397-10-325-3]](/bjoc/content/figures/1860-5397-10-325-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Viabilities of Caco-2 cells incubated with a) 5a after 2 h, b) 5a after 24 h, c) DIMEB after 2 h, d) DIMEB after 24 h, in serum (black), in basal medium (grey).
Figure 3: Viabilities of Caco-2 cells incubated with a) 5a after 2 h, b) 5a after 24 h, c) DIMEB after 2 h, d...
According to DIN EN ISO 10993-5 [51], a more than 30% deviation of measurement values of treated cells compared to the untreated control was defined as cytotoxic. After treatment of the Caco-2 cell line, not any cytotoxic effect could be detected for all of the tested concentrations of 5a after 2 h of incubation, while DIMEB was already highly toxic at 25 mg/mL. After 24 h of incubation, 5a also showed a rather weak toxicity, while DIMEB was already highly toxic at the clinically relevant concentration (see Figure 3). The effect of the culture medium was negligible.
Complexation of anaesthetic drugs by CD polymers
The complexation of the anaesthetic drug sevoflurane (shown in Scheme 4) by the methylated CDs and CD polymers was quantified by measurement of the vapor pressure of the sevoflurane by gas chromatography as a function of the CD concentration as already described previously [14,52]. The respective occupancies of the hosts occupancies (the molar ratios of complexed guest vs. host) are listed in Table 2. While native β-CD shows only a weak affinity to sevoflurane, the methylated CDs RAMEB and especially DIMEB show satisfactory occupancies. Methylation increases the hydrophobicity of the CD cavity and therefore improves the compatibility with the hydrophobic guest sevoflurane. Surprisingly, the completely methylated β-CD TRIMEB had a much lower binding potential to sevoflurane than all other CD derivatives. Low binding affinities of TRIMEB are also known towards other guest molecules, such as tert-butyl benzoate and adamantane-1-carboxylate, and had been rationalized by the lack of intramolecular hydrogen bonds which would otherwise rigidify the CD scaffold [16]. The occupancies of the HES polymers (see Table 2) also increased with increasing degree of methylation, DSCD(CH3) similar to the monomeric CDs. The anionic polymer 6 showed a somewhat smaller binding potential.
![[1860-5397-10-325-i4]](/bjoc/content/inline/1860-5397-10-325-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Structures of a) midazolam and b) sevoflurane.
Scheme 4: Structures of a) midazolam and b) sevoflurane.
The complexation of the anaesthetic drug midazolam (shown in Scheme 4) by the methylated CDs and CD polymers was quantified by the phase solubility method [53,54]. Solubility of midazolam was measured by UV spectroscopy as the function of the concentration of the CD derivative. The resulting occupancies are listed in Table 2. The value for native β-CD was quite low and in accordance with results previously obtained by 1H NMR spectroscopy [55]. Again DIMEB showed the highest value. Occupancies of the polymers were in the same order of magnitude or even higher than for the respective CD monomers (Table 2).
Conclusion
The attachment of DIMEB to a polymer backbone by the click-reaction is a very efficient way to synthesise polymeric hosts with both low toxicity and excellent binding potential. The conjugate of DIMEB and HES 5a is regarded as a good candidate for oral or parenteral delivery of hydrophobic drugs.
Experimental
Complexation of sevoflurane. The vapor pressure of sevoflurane was determined by head space gas chromatography with a Shimadzu GC-17A GC equipped with a head space unit from Shimadzu, Japan. Vials of 5 mL volume were used, the ratio between gas (V = 3.2 mL) and aqueous (V = 1.8 mL) phase was f = 1.77. Occupancies were calculated from the GC data as described previously [56].
Complexation of midazolam. An excess amount of midazolam was added to aqueous solutions containing various concentrations of CD polymers and stirred for 24 h at 25 °C. After equilibration, aliquots of the suspension were filtered through a 0.20 μm membrane cellulose filter, suitably diluted and analyzed using UV spectroscopy. The concentration of midazolam in each solution was determined by measuring its absorbance at 240 nm (ε = 21,240 M−1cm−1). The intrinsic absorption of each CD polymer was taken into account.
Cell viability was determined using CellTiter-Glo® assay (Promega, #G7571) and Caco-2 colon carcinoma cells (DSMZ #ACC-169; passage 9-17). Cells were cultured as described by the supplier, seeded with 5 × 103 cells per well in 96-well plates and tests carried out at about 80% cell confluency (~3–4 days). The assay results in cell lysis and generation of a luminescent signal proportional to the amount of ATP present, which is directly proportional to the number of cells in culture. The luminescence signal was measured in a plate reader (infinite M200, TECAN, Männedorf, Swizerland). 5a and DIMEB were each solved in standard basal medium (MEM) or human serum and cells were treated for 2 h and 24 h with the above-mentioned concentrations. For viability assessment, the test substances were removed by washing with PBS buffer (Sigma®), the cells in each well were overlaid with 100 µL of basal medium and 100 μL of CellTiter-Glo® reagent and luminescence was measured after 2 min of shaking and 10 min incubation at room temperature in the TECAN plate reader. All results presented are based on at least three independent biological tests. Statistical significance was determined by analysis of variance and p < 0.05.
Materials: Hydroxyethyl starch (HES) with an average molar mass (Mw) of 130 kDa and a molar substitution of 0.4 was kindly provided by Fresenius Kabi, Friedberg, Germany (batch no: 17120211). β-CD was obtained from Wacker Chemie GmbH, München, Germany. HES and CD were used after drying overnight at 70 °C under reduced pressure. All other reagents and solvents were purchased from commercial suppliers and used as received. Mono-6-deoxy-6-azido-β-CD was synthesized according to a literature protocol [57]. All complexation studies were performed in saline HEPES-buffer solution (pH 7.4) with a NaCl concentration of 0.9 wt %.
Mono-6-deoxy-6-azido-heptakis(2,6-di-O-methyl)-β-cyclodextrin (1a): 3.0 g (2.59 mmol) mono-(6-deoxy-6-azido)-β-CD was dissolved in 150 mL DMF/DMSO 1:1 (v/v) under N2 and a mixture of 26 g (168 mmol) BaO and 26 g Ba(OH)2 (153 mmol) was added in small portions within 10 min under stirring. The mixture was cooled to 0 °C and 30 mL (0.32 mol) dimethyl sulfate was added over a period of 1 h under intensive stirring keeping the internal temperature below 5 °C. Stirring was continued for 4 d at rt. Then the mixture was warmed up to 85 °C for 30 min. After cooling to rt, excess dimethyl sulfate was decomposed by addition of 50 mL of 25 wt % aqueous ammonia. The mixture was stirred for 16 h and neutralized by addition of conc. HCl. Then 180 mL chloroform was added, filtered and the precipitate was washed with further 80 mL chloroform. The filtrate was concentrated to dryness in vacuum, dissolved in water, purified by nanofiltration with water against a polyethersulfone membrane (cut-off: 1 kDa) and lyophilized.
Yield: 2.5 g (1.86 mmol, 72%); DS: 2.2; 1H NMR (400 MHz, CDCl3) δ 5.25–4.94 (m, 12H, H-1/1‘, OH-3/3‘), 3.94–3.92 (m, 7H, H-3/3‘), 3.75–3.40 (m, 74H, H-4/4‘, H-5/5‘, H-6/6’a/6’b, H-7/7‘, H-8, H-9), 3.29–3.21 (m, 7H, H-2/2‘); 13C NMR (100 MHz, CDCl3) δ 101.3 (C-1), 83.5 (C-4), 82.0 (C-2), 73.2 (C-3), 70.8 (C-6a/b), 70.3 (C-5), 61.7 - 58.2 (C-7, C-8, C-9), 51.8 (C-6’a/b); MS (m/z): [M + Na+] 1364.63, C55H95N3O34, (m/z): [M + Na+ + CH3] 1378.02, C56H97N3O34, (m/z): [M + Na+ + 2CH3] 1392.64, C57H99N3O34, most intense peak (m/z): [M + Na+ + 3CH3] 1406.29, C58H101N3O34, (m/z): [M + Na+ + 4CH3] 1420.20, C59H103N3O34; IR ![[Graphic 1]](/bjoc/content/inline/1860-5397-10-325-i5.svg?max-width=637&scale=1.18182) (cm−1): 3412 (OH), 2930 (CH), 2102 (N3), 1453 (CH), 1361 (OH).
(cm−1): 3412 (OH), 2930 (CH), 2102 (N3), 1453 (CH), 1361 (OH).
(3-Propargyloxy-2-hydroxypropyl)-hydroxyethyl starch (2): 3.412 g (18.98 mmol) HES was dissolved in 40 mL 0.1 M NaOH and 1.8 mL (16.70 mmol) glycidyl propargyl ether were added. The temperature was increased to 35 °C and the solution was stirred. After 24 h further 1.8 mL (16.70 mmol) glycidyl propargyl ether were added and stirred for another 16 h. The product was precipitated in 500 mL 2-propanol, filtered and washed with 200 mL 2-propanol. The product was purified by ultrafiltration with water against a polyethersulfone membrane (cut-off: 5 kDa) and freeze-dried.
Yield: 3.61 g (14.97 mmol, 79%); DS: 0.65; 1H NMR (400 MHz, DMSO-d6/D2O) δ 5.68–5.38 (m, 1H, H-01/01‘), 4.27 (s, 1.30H, H-12‘), 4.03–3.45 (m, 10H, H-02/02‘, H-03/03‘, H-04/04‘, H-05/05‘, H-06/06‘, H-07/07‘, H-08/08,H-9‘, H-10‘, H-11‘, H-14‘); IR (cm−1): 3400.25 (OH), 2925.80 (CH), 2113.83 (-C≡C), 1456.15 (CH), 1365.50 (OH).
Conjugate of DIMEB and HES 5a: Under an atmosphere of N2 1.6 g (1.19 mmol) 1a was dissolved in 40 mL of degassed DMSO/H2O 1:1 (v/v) and 600 mg (2.4 mmol) 2 and 334 µL (119 µmol) of a solution of sodium ascorbate in water (70 mg/mL) were added. After reaching 50 °C, 211 µL (59 µmol) of a solution of CuSO4·5H2O in water (70 mg/mL) was added. The solution was stirred for 48 h and purified by ultrafiltration with water against a polyethersulfone membrane (cut-off: 5 kDa) and freeze-dried.
Yield: 1.67 g (2.15 mmol, 90%); DS: 0.4; 1H NMR: (400 MHz, D2O) δ 8.03 (s, 0.4H, H-14‘), 5.58 (m, 1H, H-01/01‘), 5.14 (m, 2.8H, H-1/1‘), 3.86–3.07 (m, 45H, H-02/02‘–H-08/08‘, H2/2‘–H-7/7‘, H8, H9‘–H12‘); IR (cm−1): 3400.25 (OH), 2925.80 (CH), 1456.15 (CH), 1365.50 (OH).
Supporting Information
| Supporting Information File 1: General methods and experimental procedures for compounds 1b, 3, 4, 5b and for the oxidation of HES. | ||
| Format: PDF | Size: 104.4 KB | Download |
| Supporting Information File 2: NMR spectra of HES, 1a, 1b, 6, and IR spectra of HES, 2, 5a. | ||
| Format: PDF | Size: 333.5 KB | Download |
References
-
Szejtli, J. Pure Appl. Chem. 2004, 76, 1825–1845. doi:10.1351/pac200476101825
Return to citation in text: [1] -
Rekharsky, M. V.; Inoue, Y. Chem. Rev. 1998, 98, 1875–1918. doi:10.1021/cr970015o
Return to citation in text: [1] [2] -
Wang, W.; Kaifer, A. E. Cucurbituril and Cyclodextrin Complexes of Dendrimers. In Inclusion Polymers; Wenz, G., Ed.; Springer-Verlag: Berlin, 2009; Vol. 222, pp 1–54. doi:10.1007/12_2008_1
Return to citation in text: [1] -
Irie, T.; Uekama, K. J. Pharm. Sci. 1997, 86, 147–162. doi:10.1021/js960213f
Return to citation in text: [1] -
Kiss, T.; Fenyvesi, F.; Bácskay, I.; Váradi, J.; Fenyvesi, É.; Iványi, R.; Szente, L.; Tósaki, Á.; Vecsernyés, M. Eur. J. Pharm. Sci. 2010, 40, 376–380. doi:10.1016/j.ejps.2010.04.014
Return to citation in text: [1] [2] -
Frank, D. W.; Gray, J. E.; Weaver, R. N. Am. J. Pathol. 1976, 83, 367–382.
Return to citation in text: [1] -
Frijlink, H. W.; Eissens, A. C.; Hefting, N. R.; Poelstra, K.; Lerk, C. F.; Meijer, D. K. F. Pharm. Res. 1991, 8, 9–16. doi:10.1023/A:1015861719134
Return to citation in text: [1] -
Kimura, E.; Bersani-Amado, C. A.; Sudo, L. S.; Santos, S. R. J.; Oga, S. Gen. Pharmacol. 1997, 28, 695–698. doi:10.1016/S0306-3623(96)00362-X
Return to citation in text: [1] -
Brewster, M. E.; Loftsson, T. Adv. Drug Delivery Rev. 2007, 59, 645–666. doi:10.1016/j.addr.2007.05.012
Return to citation in text: [1] -
Bilensoy, E., Ed. Cyclodextrins in pharmaceutics, cosmetics, and biomedicine; John Wiley & Sons: Hoboken, NJ, 2011; pp 1–395. doi:10.1002/9780470926819.ins
Return to citation in text: [1] -
Loftsson, T.; Brewster, M. E. J. Pharm. Pharmacol. 2010, 62, 1607–1621. doi:10.1111/j.2042-7158.2010.01030.x
Return to citation in text: [1] -
Leroy-Lechat, F.; Wouessidjewe, D.; Andreux, J.-P.; Puisieux, F.; Duchêne, D. Int. J. Pharm. 1994, 101, 97–103. doi:10.1016/0378-5173(94)90080-9
Return to citation in text: [1] -
Zia, V.; Rajewski, R. A.; Stella, V. J. Pharm. Res. 2001, 18, 667–673. doi:10.1023/A:1011041628797
Return to citation in text: [1] -
Fourmentin, S.; Ciobanu, A.; Landy, D.; Wenz, G. Beilstein J. Org. Chem. 2013, 9, 1185–1191. doi:10.3762/bjoc.9.133
Return to citation in text: [1] [2] -
Szejtli, J.; Liptak, A.; Jodal, I.; Fügedi, P.; Nanasi, P.; Neszmelyi, A. Starch/Staerke 1980, 32, 162–164.
Return to citation in text: [1] [2] -
Wenz, G. Beilstein J. Org. Chem. 2012, 8, 1890–1895. doi:10.3762/bjoc.8.218
Return to citation in text: [1] [2] -
Szejtli, J. J. Inclusion Phenom. Macrocyclic Chem. 1983, 1, 135–150. doi:10.1007/BF00656816
Return to citation in text: [1] -
Carrier, R. L.; Miller, L. A.; Ahmed, I. J. Controlled Release 2007, 123, 78–99. doi:10.1016/j.jconrel.2007.07.018
Return to citation in text: [1] -
Challa, R.; Ahuja, A.; Ali, J.; Khar, R. K. AAPS PharmSciTech 2005, 6, R329–E357. doi:10.1208/pt060243
Return to citation in text: [1] -
Cheng, J.; Khin, K. T.; Jensen, G. S.; Liu, A.; Davis, M. E. Bioconjugate Chem. 2003, 14, 1007–1017. doi:10.1021/bc0340924
Return to citation in text: [1] -
Weickenmeier, M.; Wenz, G. Macromol. Rapid Commun. 1996, 17, 731–736. doi:10.1002/marc.1996.030171008
Return to citation in text: [1] -
Ramírez, H. L.; Valdivia, A.; Cao, R.; Torres-Labandeira, J. J.; Fragoso, A.; Villalonga, R. Bioorg. Med. Chem. Lett. 2006, 16, 1499–1501. doi:10.1016/j.bmcl.2005.12.049
Return to citation in text: [1] -
Ramirez, H. L.; Valdivia, A.; Cao, R.; Fragoso, A.; Torres Labandeira, J. J.; Baños, M.; Villalonga, R. Polym. Bull. 2007, 59, 597–605. doi:10.1007/s00289-007-0803-8
Return to citation in text: [1] -
Gomez, C. G.; Chambat, G.; Heyraud, A.; Villar, M.; Auzély-Velty, R. Polymer 2006, 47, 8509–8516. doi:10.1016/j.polymer.2006.10.011
Return to citation in text: [1] [2] -
Charlot, A.; Heyraud, A.; Guenot, P.; Rinaudo, M.; Auzély-Velty, R. Biomacromolecules 2006, 7, 907–913. doi:10.1021/bm0507094
Return to citation in text: [1] -
Aoki, N.; Nishikawa, M.; Hattori, K. Carbohydr. Polym. 2003, 52, 219–223. doi:10.1016/S0144-8617(02)00308-9
Return to citation in text: [1] -
Prabaharan, M.; Mano, J. F. Macromol. Biosci. 2005, 5, 965–973. doi:10.1002/mabi.200500087
Return to citation in text: [1] -
Nielsen, T. T.; Wintgens, V.; Amiel, C.; Wimmer, R.; Larsen, K. L. Biomacromolecules 2010, 11, 1710–1715. doi:10.1021/bm9013233
Return to citation in text: [1] [2] [3] [4] -
Yuan, Z.; Ye, Y.; Gao, F.; Yuan, H.; Lan, M.; Lou, K.; Wang, W. Int. J. Pharm. 2013, 446, 191–198. doi:10.1016/j.ijpharm.2013.02.024
Return to citation in text: [1] -
Huisgen, R.; Szeimies, G.; Möbius, L. Chem. Ber. 1967, 100, 2494–2507. doi:10.1002/cber.19671000806
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1] [2] -
Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e
Return to citation in text: [1] -
Treib, J.; Baron, J.-F.; Grauer, M. T.; Strauss, R. G. Intensive Care Med. 1999, 25, 258–268. doi:10.1007/s001340050833
Return to citation in text: [1] -
Schortgen, F.; Deye, N.; Brochard, L. Intensive Care Med. 2004, 30, 2222–2229. doi:10.1007/s00134-004-2415-1
Return to citation in text: [1] -
Boldt, J. Anesth. Analg. 2009, 108, 1574–1582. doi:10.1213/ane.0b013e31819e9e6c
Return to citation in text: [1] -
Delgado-Herrera, L.; Ostroff, R. D.; Rogers, S. A. CNS Drug Rev. 2001, 7, 48–120. doi:10.1111/j.1527-3458.2001.tb00190.x
Return to citation in text: [1] -
Gudmundsdottir, H.; Sigurjonsdottir, J. F.; Masson, M.; Fjalldal, O.; Stefansson, E.; Loftsson, T. Pharmazie 2001, 56, 963–966.
Return to citation in text: [1] -
Marcon, F.; Mathiron, D.; Pilard, S.; Lemaire-Hurtel, A.-S.; Dubaele, J.-M.; Djedaini-Pilard, F. Int. J. Pharm. 2009, 379, 244–250. doi:10.1016/j.ijpharm.2009.05.029
Return to citation in text: [1] -
Hocquelet, C.; Blu, J.; Jankowski, C. K.; Arseneau, S.; Buisson, D.; Mauclaire, L. Tetrahedron 2006, 62, 11963–11971. doi:10.1016/j.tet.2006.09.089
Return to citation in text: [1] -
Casu, B.; Reggiani, M.; Gallo, G. G.; Vigevani, A. Tetrahedron 1968, 24, 803–821. doi:10.1016/0040-4020(68)88030-5
Return to citation in text: [1] -
Wimmer, T. Preparation of alkylated cyclodextrin derivatives, methylated cyclodextrin derivatives and their use. U.S. Patent 5710268, April 5, 1994.
Return to citation in text: [1] [2] -
Gelb, R. I.; Schwartz, L. M.; Bradshaw, J. J.; Laufer, D. A. Bioorg. Chem. 1980, 9, 299–304. doi:10.1016/0045-2068(80)90039-5
Return to citation in text: [1] -
Gelb, R. I.; Schwartz, L. M.; Laufer, D. A. Bioorg. Chem. 1982, 11, 274–280. doi:10.1016/0045-2068(82)90003-7
Return to citation in text: [1] -
Augsten, C. Asymmetrische Fluß Feld-Fluß Fraktionierung in Verbindung mit Mehrwinkellichtstreudetektion – Eine neue bedeutende Methode der Pharmazeutischen Analytik zur Charakterisierung von Makromolekülen und Nanopartikeln. Ph.D. Thesis, Martin-Luther-Universität , Halle-Wittenberg, Germany, 2008.
Return to citation in text: [1] -
Thiele, C.; Auerbach, D.; Jung, G.; Qiong, L.; Schneider, M.; Wenz, G. Polym. Chem. 2011, 2, 209–215. doi:10.1039/c0py00241k
Return to citation in text: [1] -
Bragd, P. L.; Besemer, A. C.; van Bekkum, H. Carbohydr. Res. 2000, 328, 355–363. doi:10.1016/S0008-6215(00)00109-9
Return to citation in text: [1] -
Kern, H.; Choi, S.; Wenz, G.; Heinrich, J.; Ehrhardt, L.; Mischnick, P.; Garidel, P.; Blume, A. Carbohydr. Res. 2000, 326, 67–79. doi:10.1016/S0008-6215(00)00024-0
Return to citation in text: [1] -
Yan, J.; Li, W.; Zhang, X.; Liu, K.; Wu, P.; Zhang, A. J. Mater. Chem. 2012, 22, 17424–17428. doi:10.1039/c2jm33328g
Return to citation in text: [1] -
Ju, B.; Zhang, C.; Zhang, S. Carbohydr. Polym. 2014, 108, 307–312. doi:10.1016/j.carbpol.2014.02.057
Return to citation in text: [1] -
Hannah, R.; Beck, M.; Moravec, R.; Riss, T. Promega Cell Notes 2001, 2, 11–13.
Return to citation in text: [1] -
ISO 10993-5:2009(en) Biological evaluation of medical devices – Part 5: Tests for in vitro cytotoxicity. https://www.iso.org/obp/ui/#iso:std:iso:10993:-5:ed-3:v1:en.
Return to citation in text: [1] -
Lantz, A. W.; Wetterer, S. M.; Armstrong, D. W. Anal. Bioanal. Chem. 2005, 383, 160–166. doi:10.1007/s00216-005-0030-9
Return to citation in text: [1] -
Higuchi, T.; Connors, K. A. Adv. Anal. Chem. Instrum. 1965, 4, 117–212.
Return to citation in text: [1] -
Connors, K. A. Chem. Rev. 1997, 97, 1325–1358. doi:10.1021/cr960371r
Return to citation in text: [1] -
Ali, S. M.; Upadhyay, S. K. Magn. Reson. Chem. 2008, 46, 676–679. doi:10.1002/mrc.2231
Return to citation in text: [1] -
Becker, L. F.; Schwarz, D. H.; Wenz, G. Beilstein J. Org. Chem. 2014, 10, 2920–2927. doi:10.3762/bjoc.10.310
Return to citation in text: [1] -
Tang, W.; Ng, S.-C. Nat. Protoc. 2008, 3, 691–697. doi:10.1038/nprot.2008.37
Return to citation in text: [1]
| 36. | Delgado-Herrera, L.; Ostroff, R. D.; Rogers, S. A. CNS Drug Rev. 2001, 7, 48–120. doi:10.1111/j.1527-3458.2001.tb00190.x |
| 37. | Gudmundsdottir, H.; Sigurjonsdottir, J. F.; Masson, M.; Fjalldal, O.; Stefansson, E.; Loftsson, T. Pharmazie 2001, 56, 963–966. |
| 38. | Marcon, F.; Mathiron, D.; Pilard, S.; Lemaire-Hurtel, A.-S.; Dubaele, J.-M.; Djedaini-Pilard, F. Int. J. Pharm. 2009, 379, 244–250. doi:10.1016/j.ijpharm.2009.05.029 |
| 39. | Hocquelet, C.; Blu, J.; Jankowski, C. K.; Arseneau, S.; Buisson, D.; Mauclaire, L. Tetrahedron 2006, 62, 11963–11971. doi:10.1016/j.tet.2006.09.089 |
| 6. | Frank, D. W.; Gray, J. E.; Weaver, R. N. Am. J. Pathol. 1976, 83, 367–382. |
| 7. | Frijlink, H. W.; Eissens, A. C.; Hefting, N. R.; Poelstra, K.; Lerk, C. F.; Meijer, D. K. F. Pharm. Res. 1991, 8, 9–16. doi:10.1023/A:1015861719134 |
| 21. | Weickenmeier, M.; Wenz, G. Macromol. Rapid Commun. 1996, 17, 731–736. doi:10.1002/marc.1996.030171008 |
| 44. | Augsten, C. Asymmetrische Fluß Feld-Fluß Fraktionierung in Verbindung mit Mehrwinkellichtstreudetektion – Eine neue bedeutende Methode der Pharmazeutischen Analytik zur Charakterisierung von Makromolekülen und Nanopartikeln. Ph.D. Thesis, Martin-Luther-Universität , Halle-Wittenberg, Germany, 2008. |
| 4. | Irie, T.; Uekama, K. J. Pharm. Sci. 1997, 86, 147–162. doi:10.1021/js960213f |
| 5. | Kiss, T.; Fenyvesi, F.; Bácskay, I.; Váradi, J.; Fenyvesi, É.; Iványi, R.; Szente, L.; Tósaki, Á.; Vecsernyés, M. Eur. J. Pharm. Sci. 2010, 40, 376–380. doi:10.1016/j.ejps.2010.04.014 |
| 22. | Ramírez, H. L.; Valdivia, A.; Cao, R.; Torres-Labandeira, J. J.; Fragoso, A.; Villalonga, R. Bioorg. Med. Chem. Lett. 2006, 16, 1499–1501. doi:10.1016/j.bmcl.2005.12.049 |
| 23. | Ramirez, H. L.; Valdivia, A.; Cao, R.; Fragoso, A.; Torres Labandeira, J. J.; Baños, M.; Villalonga, R. Polym. Bull. 2007, 59, 597–605. doi:10.1007/s00289-007-0803-8 |
| 24. | Gomez, C. G.; Chambat, G.; Heyraud, A.; Villar, M.; Auzély-Velty, R. Polymer 2006, 47, 8509–8516. doi:10.1016/j.polymer.2006.10.011 |
| 25. | Charlot, A.; Heyraud, A.; Guenot, P.; Rinaudo, M.; Auzély-Velty, R. Biomacromolecules 2006, 7, 907–913. doi:10.1021/bm0507094 |
| 45. | Thiele, C.; Auerbach, D.; Jung, G.; Qiong, L.; Schneider, M.; Wenz, G. Polym. Chem. 2011, 2, 209–215. doi:10.1039/c0py00241k |
| 46. | Bragd, P. L.; Besemer, A. C.; van Bekkum, H. Carbohydr. Res. 2000, 328, 355–363. doi:10.1016/S0008-6215(00)00109-9 |
| 3. | Wang, W.; Kaifer, A. E. Cucurbituril and Cyclodextrin Complexes of Dendrimers. In Inclusion Polymers; Wenz, G., Ed.; Springer-Verlag: Berlin, 2009; Vol. 222, pp 1–54. doi:10.1007/12_2008_1 |
| 5. | Kiss, T.; Fenyvesi, F.; Bácskay, I.; Váradi, J.; Fenyvesi, É.; Iványi, R.; Szente, L.; Tósaki, Á.; Vecsernyés, M. Eur. J. Pharm. Sci. 2010, 40, 376–380. doi:10.1016/j.ejps.2010.04.014 |
| 18. | Carrier, R. L.; Miller, L. A.; Ahmed, I. J. Controlled Release 2007, 123, 78–99. doi:10.1016/j.jconrel.2007.07.018 |
| 19. | Challa, R.; Ahuja, A.; Ali, J.; Khar, R. K. AAPS PharmSciTech 2005, 6, R329–E357. doi:10.1208/pt060243 |
| 28. | Nielsen, T. T.; Wintgens, V.; Amiel, C.; Wimmer, R.; Larsen, K. L. Biomacromolecules 2010, 11, 1710–1715. doi:10.1021/bm9013233 |
| 2. | Rekharsky, M. V.; Inoue, Y. Chem. Rev. 1998, 98, 1875–1918. doi:10.1021/cr970015o |
| 20. | Cheng, J.; Khin, K. T.; Jensen, G. S.; Liu, A.; Davis, M. E. Bioconjugate Chem. 2003, 14, 1007–1017. doi:10.1021/bc0340924 |
| 42. | Gelb, R. I.; Schwartz, L. M.; Bradshaw, J. J.; Laufer, D. A. Bioorg. Chem. 1980, 9, 299–304. doi:10.1016/0045-2068(80)90039-5 |
| 43. | Gelb, R. I.; Schwartz, L. M.; Laufer, D. A. Bioorg. Chem. 1982, 11, 274–280. doi:10.1016/0045-2068(82)90003-7 |
| 2. | Rekharsky, M. V.; Inoue, Y. Chem. Rev. 1998, 98, 1875–1918. doi:10.1021/cr970015o |
| 14. | Fourmentin, S.; Ciobanu, A.; Landy, D.; Wenz, G. Beilstein J. Org. Chem. 2013, 9, 1185–1191. doi:10.3762/bjoc.9.133 |
| 41. | Wimmer, T. Preparation of alkylated cyclodextrin derivatives, methylated cyclodextrin derivatives and their use. U.S. Patent 5710268, April 5, 1994. |
| 13. | Zia, V.; Rajewski, R. A.; Stella, V. J. Pharm. Res. 2001, 18, 667–673. doi:10.1023/A:1011041628797 |
| 17. | Szejtli, J. J. Inclusion Phenom. Macrocyclic Chem. 1983, 1, 135–150. doi:10.1007/BF00656816 |
| 41. | Wimmer, T. Preparation of alkylated cyclodextrin derivatives, methylated cyclodextrin derivatives and their use. U.S. Patent 5710268, April 5, 1994. |
| 11. | Loftsson, T.; Brewster, M. E. J. Pharm. Pharmacol. 2010, 62, 1607–1621. doi:10.1111/j.2042-7158.2010.01030.x |
| 12. | Leroy-Lechat, F.; Wouessidjewe, D.; Andreux, J.-P.; Puisieux, F.; Duchêne, D. Int. J. Pharm. 1994, 101, 97–103. doi:10.1016/0378-5173(94)90080-9 |
| 15. | Szejtli, J.; Liptak, A.; Jodal, I.; Fügedi, P.; Nanasi, P.; Neszmelyi, A. Starch/Staerke 1980, 32, 162–164. |
| 8. | Kimura, E.; Bersani-Amado, C. A.; Sudo, L. S.; Santos, S. R. J.; Oga, S. Gen. Pharmacol. 1997, 28, 695–698. doi:10.1016/S0306-3623(96)00362-X |
| 9. | Brewster, M. E.; Loftsson, T. Adv. Drug Delivery Rev. 2007, 59, 645–666. doi:10.1016/j.addr.2007.05.012 |
| 10. | Bilensoy, E., Ed. Cyclodextrins in pharmaceutics, cosmetics, and biomedicine; John Wiley & Sons: Hoboken, NJ, 2011; pp 1–395. doi:10.1002/9780470926819.ins |
| 15. | Szejtli, J.; Liptak, A.; Jodal, I.; Fügedi, P.; Nanasi, P.; Neszmelyi, A. Starch/Staerke 1980, 32, 162–164. |
| 40. | Casu, B.; Reggiani, M.; Gallo, G. G.; Vigevani, A. Tetrahedron 1968, 24, 803–821. doi:10.1016/0040-4020(68)88030-5 |
| 29. | Yuan, Z.; Ye, Y.; Gao, F.; Yuan, H.; Lan, M.; Lou, K.; Wang, W. Int. J. Pharm. 2013, 446, 191–198. doi:10.1016/j.ijpharm.2013.02.024 |
| 26. | Aoki, N.; Nishikawa, M.; Hattori, K. Carbohydr. Polym. 2003, 52, 219–223. doi:10.1016/S0144-8617(02)00308-9 |
| 27. | Prabaharan, M.; Mano, J. F. Macromol. Biosci. 2005, 5, 965–973. doi:10.1002/mabi.200500087 |
| 31. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 28. | Nielsen, T. T.; Wintgens, V.; Amiel, C.; Wimmer, R.; Larsen, K. L. Biomacromolecules 2010, 11, 1710–1715. doi:10.1021/bm9013233 |
| 47. | Kern, H.; Choi, S.; Wenz, G.; Heinrich, J.; Ehrhardt, L.; Mischnick, P.; Garidel, P.; Blume, A. Carbohydr. Res. 2000, 326, 67–79. doi:10.1016/S0008-6215(00)00024-0 |
| 48. | Yan, J.; Li, W.; Zhang, X.; Liu, K.; Wu, P.; Zhang, A. J. Mater. Chem. 2012, 22, 17424–17428. doi:10.1039/c2jm33328g |
| 49. | Ju, B.; Zhang, C.; Zhang, S. Carbohydr. Polym. 2014, 108, 307–312. doi:10.1016/j.carbpol.2014.02.057 |
| 50. | Hannah, R.; Beck, M.; Moravec, R.; Riss, T. Promega Cell Notes 2001, 2, 11–13. |
| 34. | Schortgen, F.; Deye, N.; Brochard, L. Intensive Care Med. 2004, 30, 2222–2229. doi:10.1007/s00134-004-2415-1 |
| 35. | Boldt, J. Anesth. Analg. 2009, 108, 1574–1582. doi:10.1213/ane.0b013e31819e9e6c |
| 28. | Nielsen, T. T.; Wintgens, V.; Amiel, C.; Wimmer, R.; Larsen, K. L. Biomacromolecules 2010, 11, 1710–1715. doi:10.1021/bm9013233 |
| 55. | Ali, S. M.; Upadhyay, S. K. Magn. Reson. Chem. 2008, 46, 676–679. doi:10.1002/mrc.2231 |
| 33. | Treib, J.; Baron, J.-F.; Grauer, M. T.; Strauss, R. G. Intensive Care Med. 1999, 25, 258–268. doi:10.1007/s001340050833 |
| 56. | Becker, L. F.; Schwarz, D. H.; Wenz, G. Beilstein J. Org. Chem. 2014, 10, 2920–2927. doi:10.3762/bjoc.10.310 |
| 30. | Huisgen, R.; Szeimies, G.; Möbius, L. Chem. Ber. 1967, 100, 2494–2507. doi:10.1002/cber.19671000806 |
| 31. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 32. | Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e |
| 53. | Higuchi, T.; Connors, K. A. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. |
| 54. | Connors, K. A. Chem. Rev. 1997, 97, 1325–1358. doi:10.1021/cr960371r |
| 24. | Gomez, C. G.; Chambat, G.; Heyraud, A.; Villar, M.; Auzély-Velty, R. Polymer 2006, 47, 8509–8516. doi:10.1016/j.polymer.2006.10.011 |
| 51. | ISO 10993-5:2009(en) Biological evaluation of medical devices – Part 5: Tests for in vitro cytotoxicity. https://www.iso.org/obp/ui/#iso:std:iso:10993:-5:ed-3:v1:en. |
| 28. | Nielsen, T. T.; Wintgens, V.; Amiel, C.; Wimmer, R.; Larsen, K. L. Biomacromolecules 2010, 11, 1710–1715. doi:10.1021/bm9013233 |
| 14. | Fourmentin, S.; Ciobanu, A.; Landy, D.; Wenz, G. Beilstein J. Org. Chem. 2013, 9, 1185–1191. doi:10.3762/bjoc.9.133 |
| 52. | Lantz, A. W.; Wetterer, S. M.; Armstrong, D. W. Anal. Bioanal. Chem. 2005, 383, 160–166. doi:10.1007/s00216-005-0030-9 |
© 2014 Markenstein et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)