Abstract
A concise asymmetric synthetic route to two new tetrahydroxyindolizidinone and quinolizidinone derivatives has been developed from a common intermediate which featured a highly selective dihydroxylation reaction and a RCM reaction as key steps.

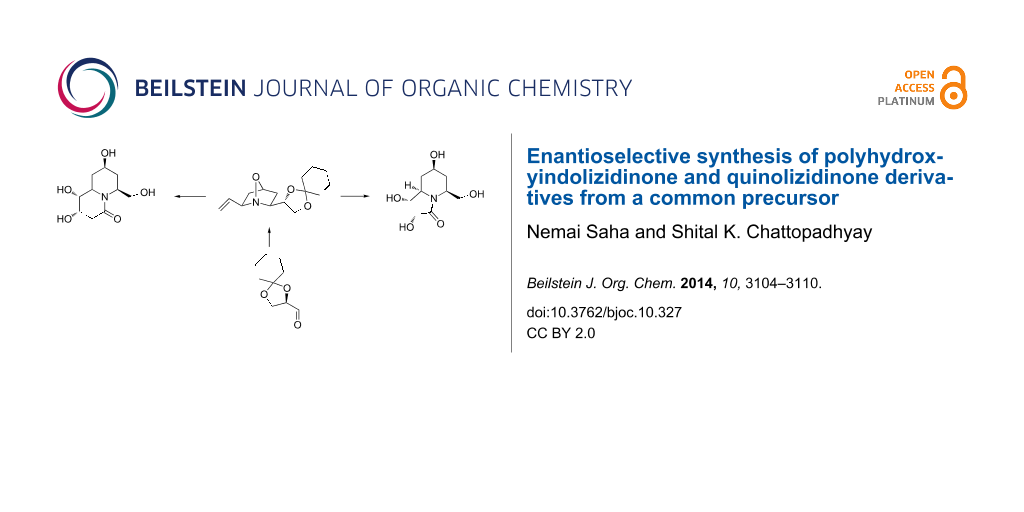
Graphical Abstract
Introduction
Polyhydroxylated indolizidine derivatives have attracted continued interest from both organic and medicinal chemists owing to their powerful biological activities [1-3]. For example, swainsonine (1, Figure 1) [4] and castanospermine (2) [5] obtained from natural sources have potent inhibitory effects towards various glycosidase enzymes and also exhibit anti-HIV, antimetastatic, immunoregulating, antitumor, and anticancer activities [6-9]. Although naturally occurring polyhydroxylated quinolizidines are less documented, several synthetic derivatives have been prepared in the quest for analogues of the more abundant indolizidines [10-15]. Ring-size variation and/or stereochemical manipulation of the hydroxy groups have been adequately practiced for such purpose [16,17]. Indolizidines and quinolizidines with fewer hydroxy groups such as lentiginosine (3) [18,19] and lupinine (4) [20] also display a useful level of biological activities. For this, and other reasons, several novel methodologies have been developed towards the synthesis of polyhydroxylated indolizidine and quinolizidine derivatives as analogues of natural products which involved RCM [21-24], dipolar cycloaddition [25,26], nucleophilic substitution [27,28], diazo insertion [29], ring expansion–transannular cyclization [30], Cope–House cyclization [31], etc. as key steps. Although great advances have been made, creation of diverse entities from a single source remains important. Herein, we report a synthetic entry to some polyhydroxylated indolizidine and quinolizidine derivatives from a common source and involving a common set of reactions.
![[1860-5397-10-327-1]](/bjoc/content/figures/1860-5397-10-327-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected polyhydroxyindolizidine and quinolizidines of importance.
Figure 1: Selected polyhydroxyindolizidine and quinolizidines of importance.
Results and Discussion
The bicyclic oxaza derivative 6 (Figure 2), previously prepared [32,33] by us from imine 5, was identified as a starting material where the built-in functionalities at the 2,6-positions were considered suitable for the stated purpose as demonstrated retro-synthetically in Figure 2. Thus, the cis-hydroxy groups in the tetrahydroxyindolizidine/quinolizidine derivative represented by the general structure I were thought to be obtainable by a substrate-controlled hydroxylation of the corresponding cycloalkene II wherein the protected 1,2-dihydroxyethyl side chain would serve as precursor of the hydroxymethyl unit in I on functional group manipulation. The bicyclic framework of the cycloalkene II was expected to be obtained from a successful RCM reaction of the N-tethered diene III which, in turn, could be prepared from amide bond formation between the amine IV and acrylic acid (for IIIa) or butenoic acid (for IIIb). The remaining hydroxy group at C-4 could possibly be generated by a reductive cleavage of the N–O bond in 6.
![[1860-5397-10-327-2]](/bjoc/content/figures/1860-5397-10-327-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Target bicyclic imino sugars Ia and Ib from a common intermediate IV.
Figure 2: Target bicyclic imino sugars Ia and Ib from a common intermediate IV.
Thus, treatment of compound 6 with Zn/AcOH proceeded well to give the all-cis-piperidine derivative 7 (Scheme 1) in very good yield. The 4-OH group in compound 7 was then protected as its TBDMS ether (8) wherein the use of TBS triflate was essential as the more conventional TBSCl was found to be ineffective. Treatment of the free amino group in 8 with neat acrylic acid provided the unsaturated amide 9 in readiness for a subsequent RCM reaction. Ring closure of 9 proceeded better in the presence of Grubbs’ second generation catalyst [34] to provide the indolizidine derivative 10 in good yield. Similarly, treatment of amine 8 with vinylacetic acid in the presence of EDC/HOBt under standard conditions proceeded smoothly to provide the N-tethered diene 11. Ring-closure of compound 11 proved to be more facile, as expected, and the quinolizidine derivative 12 was obtained in higher yield. The four step sequences 6→10 and 6→12 proceeded in overall yields of 56% and 67%, respectively.
![[1860-5397-10-327-i1]](/bjoc/content/inline/1860-5397-10-327-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (i) Zn/AcOH, rt, 1 h, 86%. (ii) TBSOTf, DIPEA, CH2Cl2, −5 °C, 1 h, 91%. (iii) Acrylic acid, EDC, HOBt, NMM, CH2Cl2, 0 °C to rt, 6 h, 96%. (iv) G-II (8 mol %), benzene, reflux, 24 h, 75%. (v) Vinyl acetic acid, EDC, HOBt, NMM, CH2Cl2, 0 °C to rt, 10 h, 90%. (vi) G-II (3 mol %), benzene, 50 °C, 2 h, 95%.
Scheme 1: Reagents and conditions: (i) Zn/AcOH, rt, 1 h, 86%. (ii) TBSOTf, DIPEA, CH2Cl2, −5 °C, 1 h, 91%. (i...
Having secured quick access to the unsaturated indolizidinone and quinolizidinone ring systems 10 and 12, we considered their conversion to the desired polyhydroxylated targets through dihydroxylation of the double bond. Pleasingly, dihydroxylation of compound 10 proceeded well under Upjohn conditions [35] and provided a single isomer 13 (Scheme 2) in high yield (96%). The high selectivity in the dihydroxylation step is noteworthy as in similar situations mixture of diastereomers has occasionally been formed [36,37].
![[1860-5397-10-327-i2]](/bjoc/content/inline/1860-5397-10-327-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (i) OsO4, NMO, acetone/water, rt, 12 h, 96%. (ii) NaH, THF, BnBr, Bu4NI, 0 °C to rt, 6 h, 70%. (iii) Ac2O, pyridine, rt, 12 h, 80%. (iv) HCl (2 N), THF, 18 h, 89%. (v) NaIO4, CH3CN/H2O, 5–10 °C, 30 min, (vi) NaBH4, MeOH, 0°C to rt, 30 min, 92% over two steps. (vii) H2, Pd(OH)2/C, MeOH, 3 h, 81%.
Scheme 2: Reagents and conditions: (i) OsO4, NMO, acetone/water, rt, 12 h, 96%. (ii) NaH, THF, BnBr, Bu4NI, 0...
The stereochemical identity of the newly formed stereogenic centres in 13 could not be ascertained at this stage due to the lack of well-resolved NMR data. To this end, the corresponding O-benzylated derivative 14 and the O-acetyl derivative 15 were prepared. Disappointingly, compound 14 proved to be of no advantage in this regard. On the contrary, the diacetyl derivative 15 revealed interesting 1H NMR and NOESY data which are summarized in Figure 3 (A and B).
![[1860-5397-10-327-3]](/bjoc/content/figures/1860-5397-10-327-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected nOe correlations (A) and coupling constants (B) for compound 15.
Figure 3: Selected nOe correlations (A) and coupling constants (B) for compound 15.
A strong nOe between the protons 8-H and 1-H (A), 5-H and 8a-H, 1-H and 2-H as well as the absence of a nOe between 8a-H and 1-H led us to conclude that dihydroxylation has taken place from the α-face as expected. The 1H,1H COSY experiment (Figure 4) further revealed that the absence of the correlation between the protons 1-H and 8a-H indicating a bisecting dihedral angle and hence a coupling between these two protons in the 1H NMR was not observed. These data clearly established the stereochemistry of 15 as depicted.
![[1860-5397-10-327-4]](/bjoc/content/figures/1860-5397-10-327-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H,1H COSY spectrum of compound 15.
Figure 4: 1H,1H COSY spectrum of compound 15.
The O-benzylated compound 14, however, proved to be more useful in the subsequent synthetic sequence. Thus, HCl-mediated deprotection of the acetal unit in 14 resulted in simultaneous removal of the silyl protecting group leading to the triol derivative 16 in an impressive yield of 89%. One-pot oxidative cleavage of the vicinal diol unit in the latter to a formyl group (not isolated) followed by its in situ reduction with NaBH4 delivered the hydroxymethyl chain in 17. Hydrogenolytic removal of the two benzyl ether functionalities with Pearlman’s catalyst then afforded the tetrahydroxyindolizidine derivative 18.
Similarly, in an effort towards the preparation of tetrahydroxyquinolizidine derivatives, we considered dihydroxylation of the unsaturated quinolizidine derivative 12. Pleasingly, dihydroxylation of 12 proved to be more facile and rewarding as it also led to the formation of a single isomer 19 (95% yield, Scheme 3). Repetition of the synthetic sequence on 19 detailed for the conversion 13→18, i.e., protection of the diol as its dibenzylic ether 20, acid-mediated one-pot deprotection of the acetal and silyl moieties leading to the triol 21, redox manipulation of the vicinal diol unit in the latter to a hydroxymethyl unit, and subsequent debenzylation of the resulting 22 led to the desired tetrahydroxyquinolizidine derivative 23 in an overall yield of 45% over six steps. Similarly, the pentahydroxylated quinolizidine derivative 24 was prepared from the triol 21 in view of the importance of such compounds having a dihydroxyethyl side chain.
![[1860-5397-10-327-i3]](/bjoc/content/inline/1860-5397-10-327-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: (i) OsO4, NMO, acetone/water, 6 h, 95%. (ii) NaH, THF, BnBr, Bu4NI, 0 °C to rt, 6 h, 82%. (iii) HCl (2 N), THF, 12 h, 80%. (iv) NaIO4, CH3CN/H2O, 5–10 °C, 30 min; (v) NaBH4, MeOH, 0 °C to rt, 30 min, 90% over two steps. (vi) H2/Pd(OH)2-C, MeOH, 6 h, 80% for 23 and 85% for 24.
Scheme 3: Reagents and conditions: (i) OsO4, NMO, acetone/water, 6 h, 95%. (ii) NaH, THF, BnBr, Bu4NI, 0 °C t...
The stereochemistry of the dihydroxylation reaction could not be adequately confirmed from NMR-spectroscopic measurements on 19. However, corresponding data on 23 revealed distinct coupling patterns in 3-H (δ 2.59, dd, J = 17.4, 6.6 Hz; δ 2.68 dd, J = 17.4, 4.8 Hz) protons as well as strong nOe between the protons on 6-H and 9a-H, 6-H and 8-H, 8-H and 9a-H, 1-H and 2-H and 2-H and 3-H as indicated in Figure 5. These studies led us to believe [38] the molecular conformation of 23 to be 5C8.
![[1860-5397-10-327-5]](/bjoc/content/figures/1860-5397-10-327-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Selected nOe correlations and part NOESY spectrum of compound 23 in D2O (600 MHz).
Figure 5: Selected nOe correlations and part NOESY spectrum of compound 23 in D2O (600 MHz).
Conclusion
In conclusion, we have developed an efficient synthetic route to prepare polyhydroxylated indolizidinone and quinolizidinone derivatives of potential importance, 18, 23 and 24 in overall yields of 25, 30 and 35%, respectively, from a single source in a linear sequence of nine steps. The methodology developed is a simple and concise one and hence may complement to those existing in the literature. The prepared compounds may also prove to be biologically important.
Supporting Information
| Supporting Information File 1: Experimental details and analytical data of all new compounds as well as their 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Asano, N. Glycobiology 2003, 13, 93R–104R. doi:10.1093/glycob/cwg090
Return to citation in text: [1] -
Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k
Return to citation in text: [1] -
Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. J.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017
Return to citation in text: [1] -
Colegate, S. M.; Dorling, P. R.; Huxtable, C. R. Aust. J. Chem. 1979, 32, 2257–2264. doi:10.1071/CH9792257
Return to citation in text: [1] -
Hohenschutz, L. D.; Bell, E. A.; Jewess, P. J.; Leworthy, D. P.; Pryce, R. J.; Arnold, E.; Clardy, J. Phytochemistry 1981, 20, 811–814. doi:10.1016/0031-9422(81)85181-3
Return to citation in text: [1] -
Asano, N.; Kato, A.; Watson, A. A. Mini-Rev. Med. Chem. 2001, 1, 145–154. doi:10.2174/1389557013407052
Return to citation in text: [1] -
Jacob, G. S. Curr. Opin. Struct. Biol. 1995, 5, 605–611. doi:10.1016/0959-440X(95)80051-4
Return to citation in text: [1] -
Durantel, D.; Alotte, C.; Zoulim, F. Curr. Opin. Investig. Drugs 2007, 8, 125–129.
Return to citation in text: [1] -
Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0
Return to citation in text: [1] -
Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078
Return to citation in text: [1] -
Gómez, L.; Garrabou, X.; Joglar, J.; Bujons, J.; Parella, T.; Vilaplana, C.; Cardona, P. J.; Clapés, P. Org. Biomol. Chem. 2012, 10, 6309–6321. doi:10.1039/c2ob25943e
Return to citation in text: [1] -
Tite, T.; Jacquelin, F.; Bischoff, L.; Fruit, C.; Marsais, F. Tetrahedron: Asymmetry 2010, 21, 2032–2036. doi:10.1016/j.tetasy.2010.05.036
Return to citation in text: [1] -
Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048
Return to citation in text: [1] -
Lesma, G.; Colombo, A.; Landoni, N.; Sacchetti, A.; Silvani, A. Tetrahedron: Asymmetry 2007, 18, 1948–1954. doi:10.1016/j.tetasy.2007.07.017
Return to citation in text: [1] -
Patil, N. T.; Tilekar, J. N.; Dhavale, D. D. J. Org. Chem. 2001, 66, 1065–1074. doi:10.1021/jo0010476
Return to citation in text: [1] -
Lahiri, R.; Ansari, A. A.; Vankar, Y. D. Chem. Soc. Rev. 2013, 42, 5102–5118. doi:10.1039/c3cs35525j
Return to citation in text: [1] -
Dragutan, I.; Dragutan, V.; Mitan, C.; Vosloo, H. C. M.; Delaude, L.; Demonceau, A. Beilstein J. Org. Chem. 2011, 7, 699–716. doi:10.3762/bjoc.7.81
Return to citation in text: [1] -
Prasad, K. R.; Pawar, A. B. ARKIVOC 2010, No. vi, 39–46.
And the references cited therein.
Return to citation in text: [1] -
Chandrasekhar, S.; Vijaykumar, B. V. D.; Pratap, T. V. Tetrahedron: Asymmetry 2008, 19, 746–750. doi:10.1016/j.tetasy.2008.02.017
And references cited therein.
Return to citation in text: [1] -
Hajri, M.; Blondelle, C.; Martinez, A.; Vasse, J.-L.; Szymoniak, J. Tetrahedron Lett. 2013, 54, 1029–1031. doi:10.1016/j.tetlet.2012.12.073
And references cited therein.
Return to citation in text: [1] -
Gómez-SanJuan, A.; Sotomayor, N.; Lete, E. Eur. J. Org. Chem. 2013, 6722–6732. doi:10.1002/ejoc.201300889
Return to citation in text: [1] -
Malik, M.; Witkowski, G.; Ceborska, M.; Jarosz, S. Org. Lett. 2013, 15, 6214–6217. doi:10.1021/ol403063v
Return to citation in text: [1] -
Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. J. Org. Chem. 2005, 70, 6552–6555. doi:10.1021/jo0509408
Return to citation in text: [1] -
Song, L.; Duesler, E. N.; Mariano, P. S. J. Org. Chem. 2004, 69, 7284–7293. doi:10.1021/jo040226a
Return to citation in text: [1] -
Jasiński, M.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 442–454. doi:10.1002/ejoc.201301406
Return to citation in text: [1] -
Mironiuk-Puchalska, E.; Rowicki, T.; Sas, W.; Koszytkowska-Stawińska, M. Tetrahedron 2013, 69, 9826–9831. doi:10.1016/j.tet.2013.09.008
Return to citation in text: [1] -
Zheng, J.-F.; Chen, W.; Huang, S.-Y.; Ye, J.-L.; Huang, P.-Q. Beilstein J. Org. Chem. 2007, 3, No. 41. doi:10.1186/1860-5397-3-41
Return to citation in text: [1] -
Azzouz, R.; Fruit, C.; Bischoff, L.; Marsais, F. J. Org. Chem. 2008, 73, 1154–1157. doi:10.1021/jo702141b
Return to citation in text: [1] -
Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926–9931. doi:10.1021/jo301967w
Return to citation in text: [1] -
Yun, H.; Kim, J.; Sim, J.; Lee, S.; Han, Y. T.; Chang, D.-J.; Kim, D.-D.; Suh, Y.-G. J. Org. Chem. 2012, 77, 5389–5393. doi:10.1021/jo300309z
Return to citation in text: [1] -
Zhang, W.; Sato, K.; Kato, A.; Jia, Y.-M.; Hu, X.-G.; Wilson, F. X.; van Well, R.; Horne, G.; Fleet, G. W. J.; Nash, R. J.; Yu, C.-Y. Org. Lett. 2011, 13, 4414–4417. doi:10.1021/ol201749c
Return to citation in text: [1] -
Saha, N.; Biswas, T.; Chattopadhyay, S. K. Org. Lett. 2011, 13, 5128–5131. doi:10.1021/ol2019967
Return to citation in text: [1] -
Saha, N.; Chattopadhyay, S. K. J. Org. Chem. 2012, 77, 11056–11063. doi:10.1021/jo3019329
Return to citation in text: [1] -
Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d
Return to citation in text: [1] -
VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973–1976. doi:10.1016/S0040-4039(00)78093-2
Return to citation in text: [1] -
Bataille, C. J. R.; Donohoe, T. J. Chem. Soc. Rev. 2011, 40, 114–128. doi:10.1039/b923880h
Return to citation in text: [1] -
Baumann, D.; Bennis, K.; Ripoche, I.; Théry, V.; Troin, Y. Eur. J. Org. Chem. 2008, 5289–5300. doi:10.1002/ejoc.200800684
Return to citation in text: [1] -
Belostotskii, A. M.; Markevich, E. J. Org. Chem. 2003, 68, 3055–3063. doi:10.1021/jo0266691
See for a detailed analysis of conformational preference of quinolizidine and indolizidine derivatives.
Return to citation in text: [1]
| 38. |
Belostotskii, A. M.; Markevich, E. J. Org. Chem. 2003, 68, 3055–3063. doi:10.1021/jo0266691
See for a detailed analysis of conformational preference of quinolizidine and indolizidine derivatives. |
| 35. | VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973–1976. doi:10.1016/S0040-4039(00)78093-2 |
| 36. | Bataille, C. J. R.; Donohoe, T. J. Chem. Soc. Rev. 2011, 40, 114–128. doi:10.1039/b923880h |
| 37. | Baumann, D.; Bennis, K.; Ripoche, I.; Théry, V.; Troin, Y. Eur. J. Org. Chem. 2008, 5289–5300. doi:10.1002/ejoc.200800684 |
| 1. | Asano, N. Glycobiology 2003, 13, 93R–104R. doi:10.1093/glycob/cwg090 |
| 2. | Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k |
| 3. | Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. J.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017 |
| 10. | Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078 |
| 11. | Gómez, L.; Garrabou, X.; Joglar, J.; Bujons, J.; Parella, T.; Vilaplana, C.; Cardona, P. J.; Clapés, P. Org. Biomol. Chem. 2012, 10, 6309–6321. doi:10.1039/c2ob25943e |
| 12. | Tite, T.; Jacquelin, F.; Bischoff, L.; Fruit, C.; Marsais, F. Tetrahedron: Asymmetry 2010, 21, 2032–2036. doi:10.1016/j.tetasy.2010.05.036 |
| 13. | Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048 |
| 14. | Lesma, G.; Colombo, A.; Landoni, N.; Sacchetti, A.; Silvani, A. Tetrahedron: Asymmetry 2007, 18, 1948–1954. doi:10.1016/j.tetasy.2007.07.017 |
| 15. | Patil, N. T.; Tilekar, J. N.; Dhavale, D. D. J. Org. Chem. 2001, 66, 1065–1074. doi:10.1021/jo0010476 |
| 32. | Saha, N.; Biswas, T.; Chattopadhyay, S. K. Org. Lett. 2011, 13, 5128–5131. doi:10.1021/ol2019967 |
| 33. | Saha, N.; Chattopadhyay, S. K. J. Org. Chem. 2012, 77, 11056–11063. doi:10.1021/jo3019329 |
| 6. | Asano, N.; Kato, A.; Watson, A. A. Mini-Rev. Med. Chem. 2001, 1, 145–154. doi:10.2174/1389557013407052 |
| 7. | Jacob, G. S. Curr. Opin. Struct. Biol. 1995, 5, 605–611. doi:10.1016/0959-440X(95)80051-4 |
| 8. | Durantel, D.; Alotte, C.; Zoulim, F. Curr. Opin. Investig. Drugs 2007, 8, 125–129. |
| 9. | Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0 |
| 34. | Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100–110. doi:10.1021/ja952676d |
| 5. | Hohenschutz, L. D.; Bell, E. A.; Jewess, P. J.; Leworthy, D. P.; Pryce, R. J.; Arnold, E.; Clardy, J. Phytochemistry 1981, 20, 811–814. doi:10.1016/0031-9422(81)85181-3 |
| 30. | Yun, H.; Kim, J.; Sim, J.; Lee, S.; Han, Y. T.; Chang, D.-J.; Kim, D.-D.; Suh, Y.-G. J. Org. Chem. 2012, 77, 5389–5393. doi:10.1021/jo300309z |
| 4. | Colegate, S. M.; Dorling, P. R.; Huxtable, C. R. Aust. J. Chem. 1979, 32, 2257–2264. doi:10.1071/CH9792257 |
| 31. | Zhang, W.; Sato, K.; Kato, A.; Jia, Y.-M.; Hu, X.-G.; Wilson, F. X.; van Well, R.; Horne, G.; Fleet, G. W. J.; Nash, R. J.; Yu, C.-Y. Org. Lett. 2011, 13, 4414–4417. doi:10.1021/ol201749c |
| 21. | Gómez-SanJuan, A.; Sotomayor, N.; Lete, E. Eur. J. Org. Chem. 2013, 6722–6732. doi:10.1002/ejoc.201300889 |
| 22. | Malik, M.; Witkowski, G.; Ceborska, M.; Jarosz, S. Org. Lett. 2013, 15, 6214–6217. doi:10.1021/ol403063v |
| 23. | Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. J. Org. Chem. 2005, 70, 6552–6555. doi:10.1021/jo0509408 |
| 24. | Song, L.; Duesler, E. N.; Mariano, P. S. J. Org. Chem. 2004, 69, 7284–7293. doi:10.1021/jo040226a |
| 27. | Zheng, J.-F.; Chen, W.; Huang, S.-Y.; Ye, J.-L.; Huang, P.-Q. Beilstein J. Org. Chem. 2007, 3, No. 41. doi:10.1186/1860-5397-3-41 |
| 28. | Azzouz, R.; Fruit, C.; Bischoff, L.; Marsais, F. J. Org. Chem. 2008, 73, 1154–1157. doi:10.1021/jo702141b |
| 20. |
Hajri, M.; Blondelle, C.; Martinez, A.; Vasse, J.-L.; Szymoniak, J. Tetrahedron Lett. 2013, 54, 1029–1031. doi:10.1016/j.tetlet.2012.12.073
And references cited therein. |
| 29. | Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926–9931. doi:10.1021/jo301967w |
| 18. |
Prasad, K. R.; Pawar, A. B. ARKIVOC 2010, No. vi, 39–46.
And the references cited therein. |
| 19. |
Chandrasekhar, S.; Vijaykumar, B. V. D.; Pratap, T. V. Tetrahedron: Asymmetry 2008, 19, 746–750. doi:10.1016/j.tetasy.2008.02.017
And references cited therein. |
| 16. | Lahiri, R.; Ansari, A. A.; Vankar, Y. D. Chem. Soc. Rev. 2013, 42, 5102–5118. doi:10.1039/c3cs35525j |
| 17. | Dragutan, I.; Dragutan, V.; Mitan, C.; Vosloo, H. C. M.; Delaude, L.; Demonceau, A. Beilstein J. Org. Chem. 2011, 7, 699–716. doi:10.3762/bjoc.7.81 |
| 25. | Jasiński, M.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 442–454. doi:10.1002/ejoc.201301406 |
| 26. | Mironiuk-Puchalska, E.; Rowicki, T.; Sas, W.; Koszytkowska-Stawińska, M. Tetrahedron 2013, 69, 9826–9831. doi:10.1016/j.tet.2013.09.008 |
© 2014 Saha and Chattopadhyay; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)