Abstract
The syntheses of palmitic acids and a nonadecane are reported with CF2 groups located 1,3 or 1,4 to each other along the aliphatic chain. Specifically 8,8,10,10- and 8,8,11,11-tetrafluorohexadecanoic acids (6b and 6c) are prepared as well as the singly modified analogue 8,8-difluorohexadecanoic acid (6a). Also 8,8,11,11-tetrafluorononadecane (27) is prepared as a pure hydrocarbon containing a 1,4-di-CF2 motif. The modified palmitic acids are characterized by differential scanning calorimetry (DSC) to determine melting points and phase behaviour relative to palmitic acid (62.5 °C). It emerges that 6c, with the CF2 groups placed 1,4- to each other, has a significantly higher melting point (89.9 °C) when compared to the other analogues and palmitic acid itself. It is a crystalline compound and the structure reveals an extended anti-zig-zag chain. Similarly 8,8,11,11-tetrafluorononadecane (27) adopts an extended anti-zig-zag structure. This is rationalized by dipolar relaxation between the two CF2 groups placed 1,4 to each other in the extended anti-zig-zag chain and suggests a design modification for long chain aliphatics which can introduce conformational stability.

Graphical Abstract
Introduction
The selective replacement of hydrogen by fluorine is widely practised in bio-organic and medicinal chemistry [1-4]. It is generally perceived that fluorine exerts only a moderate steric influence relative to hydrogen in organic compounds, but that the electronegativity of fluorine can have significant electronic influences [5]. The difluoromethylene (CF2) functionality has received considerably less attention as a functional group for modifying the properties of organic molecules, relative to –F and –CF3 groups. However we have recently become interested in the CF2 group, and in particular have noticed that the replacement of the two hydrogen atoms of a methylene by two fluorine atoms leads to widening of the C–CF2–C angle (~118°) and a narrowing of the F–C–F angle (104°) relative to tetrahederal geometry [6,7]. This deviation of classical sp3, towards sp2 hybridisation, imparts certain properties to the CF2 group in that it can accommodate angle strain. For example CF2 compounds display an apparent Thorpe–Ingold effect relative to CH2 in ring closing metathesis reactions (RCM) to cycloheptene [8]. Comparison of the rates of reaction with different substituents at the C-5 position of the diene precursors 1a–d, revealed that the CF2 substituent in 1c was as effective as the dicarboxylate 1a or ketal 1b in promoting RCM (Figure 1). This is attributed to C–CF2–C angle widening, which absorbs angle strain in the resultant cycloheptene 2c.
![[1860-5397-10-4-1]](/bjoc/content/figures/1860-5397-10-4-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The CF2 group in 1c accelerates RCM reactions relative to CHF (1d) and CH2 (1e) and with a similar rate to classical or Thorpe–Ingold substituents such as the ketal 1a and dicarboxylate ester 1b [8].
Figure 1: The CF2 group in 1c accelerates RCM reactions relative to CHF (1d) and CH2 (1e) and with a similar ...
In another study we have prepared cyclododecanes 3–5 with regiospecific placement of two CF2 groups around the ring [6] (Figure 2). X-ray structures reveal that the CF2 groups only ever occupy corner locations. This is a result of several factors including C–CF2–C angle widening, which relaxes 1,4-torsional strain across corner positions, lengthening the contact distance between those H(1)···H(4) interactions relative to those with CH2 at the corner. Also if the CF2 locates at an edge this would require that a C–F bond project into the middle of the ring. The larger steric influence of the fluorine, projecting into the tightly packed arrangement of endo orientated hydrogen atoms, raises the energy of such conformations. For cyclododecane, placing the CF2 groups 1,4 (3) or 1,7 (4) to each other, stabilizes the [3.3.3.3] square like conformation of the ring. However if the CF2 groups are placed 1,6 to each other as in 5, this introduces considerable distortion of the ring conformation as shown in Figure 2, because the CF2 avoids an edge location, which would place a fluorine atom endo and unfavourably into the centre of the ring.
![[1860-5397-10-4-2]](/bjoc/content/figures/1860-5397-10-4-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structures of a) 1,1,4,4- (3) b) 1,1,7,7- (4) and c) 1,1,6,6- (5) tetrafluorocyclododecanes. The CF2 groups locate at the corners, even for 5 which gives rise to a distorted ring conformation [6,7].
Figure 2: X-ray structures of a) 1,1,4,4- (3) b) 1,1,7,7- (4) and c) 1,1,6,6- (5) tetrafluorocyclododecanes. ...
As part of an on-going interest in the behaviour and influence of the CF2 group we have now explored the effect of locating two CF2 groups along an extended aliphatic chain. Long chain fatty acids present tractable model systems as they are solid materials and their physical properties are well described [9]. In this study we selected the three palmitic acid analogues 6a–c shown in Figure 3, as targets for synthesis and comparative analysis.
![[1860-5397-10-4-3]](/bjoc/content/figures/1860-5397-10-4-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Synthesis targets: Palmitic acid analogues 6a–c.
Figure 3: Synthesis targets: Palmitic acid analogues 6a–c.
Palmitic acid 6a containing a single CF2 group at C-8 was prepared as a control compound. The location for CF2 substitution in the middle of the aliphatic chain was selected as it is sufficiently remote from the carboxylic acid head group to have any electronic influence. Two additional analogues 6b and 6c were prepared, each with two CF2 groups, located 1,3 and 1,4 from each other respectively. These targets were designed to explore the significance on properties and chain stability of co-locating the CF2 groups at different distances from each other.
Results and Discussion
Synthesis of the palmitic acids 6a–c
As a general strategy palmitic acids 6a–c were prepared by aryl oxidation of long chain pentadecabenzenes [10,11]. The introduction of the CF2 groups was carried out by treatment of the appropriate precursor ketone with diethylaminosulfur trifluoride (DAST) [12,13]. The synthesis of palmitic acid 6a is illustrated in Scheme 1. At the outset aldehyde 8 was condensed with the acetylide of 1-octyne to afford propargylic alcohol 9, an alcohol which was readily oxidized to ketone 10. Treatment with DAST afforded difluoromethyleneacetylene 11 in good yield. The fluorination of propargylic ketones, to generate difluoromethyleneacetylenes, is methodology developed by Grée et al. [14-18] and it proved to be very reliable in our hands. An efficient hydrogenation generated the C-8 substituted difluoromethylenepentadecabenzene 12. Finally biphasic ruthenium tetroxide-catalyzed aryl oxidation gave the palmitic acid 6a in 24% overall yield as illustrated in Scheme 1 [10,11].
![[1860-5397-10-4-i1]](/bjoc/content/inline/1860-5397-10-4-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis route to 8,8-difluorohexadecanoic acid (6a).
Scheme 1: Synthesis route to 8,8-difluorohexadecanoic acid (6a).
For palmitic acid 6b, it was required to introduce the CF2 groups 1,3 to each other. This was achieved by sequential preparation of appropriate precursor ketones as illustrated in Scheme 2. For the first CF2 group ketone 14 was treated with DAST. Conversion to the CF2 group occurred in modest (45%) yield. Generally aliphatic ketones are less efficiently converted to CF2 groups with DAST in comparison to propargylic ketones. Progression of the resultant CF2 containing olefin 15 by epoxidation, chain extension and then oxidation, to ketone 18, generated the second fluorination substrate of the synthesis. DAST treatment gave pentadecabenzene 19, which was again oxidised by RuO3 to the corresponding palmitic acid 6b.
![[1860-5397-10-4-i2]](/bjoc/content/inline/1860-5397-10-4-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of palmitic acid analogues 6b and 6c.
Scheme 2: The synthesis of palmitic acid analogues 6b and 6c.
Palmitic acid 6c was prepared again relying on the methodology developed by Grée et al. [14-17] for introduction of the CF2 groups. Thus treatment of ketone 21 with DAST resulted in an efficient conversion to difluoromethyleneacetylene 22. This terminal acetylene is amenable to acetylide formation on treatment with BuLi [19,20] and condensation with hexaldehyde gave propargylic alcohol 23. The lithium methylenedifluoroacetylide (RCF2C≡CLi) reaction to form a C–C bond, provides a particularly useful synthon to access this 1,4-di-CF2 motif. Oxidation and then treatment of the resultant ketone 24, with DAST generated the tetrafluoroacetylene 25. Complete hydrogenation of the triple bond proved efficient and the resultant tetrafluoropentadecabenzene 26 was readily oxidized to palmitic acid 6c as illustrated in Scheme 2. This completed the syntheses of the palmitic acid analogues 6a–c.
Differential scanning calorimetry (DSC) data was then measured for all three of the palmitic acid samples 6a–c over a temperature range of −150 to 400 °C. In this way accurate melting point values were obtained. The melting point of C-8 difluorinated palmitic acid 6a (62.9 °C) was very similar to the natural palmitic acid (62.5 °C), Thus a single CF2 substitution, certainly at this location, has very little influence on the melting point. For palmitic acid 6b, with the two CF2-groups placed 1,3 to each other, the melting point (69 °C) is also similar to palmitic acid, but the phase behaviour is more complex as evidenced by the broad DSC profiles. This palmitic acid 6b was amorphous in nature and was not a crystalline solid, unlike the other two analogues 6a and 6c which formed crystals (Figure 4).
![[1860-5397-10-4-4]](/bjoc/content/figures/1860-5397-10-4-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: DSC traces for the three palmitic acid analogues 6a–c.
Figure 4: DSC traces for the three palmitic acid analogues 6a–c.
The tetrafluorinated palmitic acid 6c, with the CF2 groups located 1,4 from each other displays a sharp and significantly higher melting point (89.9 °C) than the other two palmitic acids 6a and 6b.
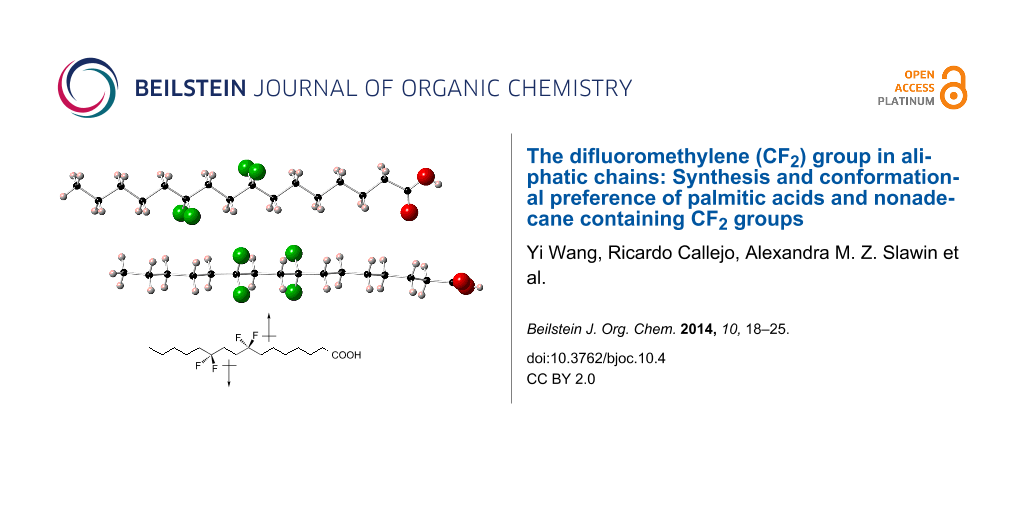
Palmitic acids 6a and 6c were crystalline solids and single crystal X-ray diffraction data were obtained for these compounds. As described above analogue 6b was amorphous in nature and despite considerable effort a single crystal could not be obtained for 6b. The resultant structures for 6a and 6c are shown in Figure 5 and Figure 6 respectively. In each case two molecules as they appear within the unit cell are presented in the image, allowing a view from above and to the side of the extended chain. The closest CF···HC contacts are 2.88 Å in 6a and 2.85 Å in 6c, much longer than any meaningful organic fluorine hydrogen bond [21]. The C–CF2–C angle in 6a (Figure 5) is 117° and as expected, wider than the other C–CH2–C angles which are typically ~112.5°. For 6c (Figure 6) the C–CF2–C angles are 115.6° (at C-8) and 116.3° (at C-11) also consistently wider that the aliphatic C–CH2–C angles. The significantly higher melting point and good crystallinity of 6c can be attributed to the relative orientation of the two CF2 groups. They are pointing perfectly anti-parallel to each other such that their dipoles cancel out in the extended anti-zig-zag chain conformation. We are currently exploring if this is a special situation whereby CF2 groups positioned 1,4 from each other can add conformational stability to aliphatic chains in other systems.
![[1860-5397-10-4-5]](/bjoc/content/figures/1860-5397-10-4-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The X-ray crystal structures of 8,8-difluorohexadecanoic acid (6a).
Figure 5: The X-ray crystal structures of 8,8-difluorohexadecanoic acid (6a).
![[1860-5397-10-4-6]](/bjoc/content/figures/1860-5397-10-4-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The X-ray structure of 8,8,11,11-tetrafluorohexadecanoic acid (6c).
Figure 6: The X-ray structure of 8,8,11,11-tetrafluorohexadecanoic acid (6c).
It occurred to us that the interactions of the carboxylate groups in palmitic acid 6c, may be dictating overall stability and conformation of the alkyl chain in the solid state. Thus it appeared appropriate to prepare a true hydrocarbon chain to further investigate the conformational preference of the 1,4-di-CF2 motif. Accordingly we selected to prepare tetrafluorononadecane 27. This is a long chain hydrocarbon with the 1,4-di-CF2 motif placed centrally. The synthetic route to 27 is illustrated in Scheme 3. The strategy for incorporating the two CF2 groups followed that used for the preparation of palmitic acid 6c. In this case propargylic ketone 30 was treated with DAST to generate difluoroacetylene 31. The resultant acetylene could then be deprotonated for conjugation to aldehyde 32. Oxidation and then fluorination of ketone 34 with DAST, introduced the second CF2 group and generated tetrafluoroacetylene 35. Finally hydrogenation of the central acetylene group gave the saturated tetrafluorononadecane 27. This compound proved to be a crystalline solid (mp 35–37 °C) with a melting point very similar to nonadecane (32–35 °C). A suitable crystal was subject to X-ray structure analysis and the resultant structure is shown in Figure 7. It is clear that the alkyl chain of 27 is extended in a similar conformation to that found in palmitic acid 6c and we conclude that this is the preferred conformation of this motif in a hydrocarbon chain.
![[1860-5397-10-4-i3]](/bjoc/content/inline/1860-5397-10-4-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis route to the tetrafluorinated alkane 27.
Scheme 3: Synthesis route to the tetrafluorinated alkane 27.
![[1860-5397-10-4-7]](/bjoc/content/figures/1860-5397-10-4-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The X-ray structure of 8,8,11,11-tetrafluorononadecane (27).
Figure 7: The X-ray structure of 8,8,11,11-tetrafluorononadecane (27).
Conclusion
In conclusion, we have synthesised three palmitic acid analogues 6a–c carrying regiospecifically located CF2 groups. The tetrafluorononadecane 27 was also prepared as an example of a true hydrocarbon. Relatively efficient synthesis protocols were devised for placing the CF2 groups 1,3 and 1,4 to each other. The CF2 groups of 6b, 6c and 27 were introduced sequentially from appropriate precursor ketones, using DAST. In particular, the methodology of Grée et al., enabled the efficient introduction of CF2 groups from propargylic ketones in the syntheses of 6a, 6c and 27. A useful C–C bond forming reaction involved a lithium methylenedifluoroacetylide (RCF2C≡CLi) condensation with an aldehyde, offers an efficient strategy for the preparation of the 1,4-di-CF2 motif after suitable functional group manipulations.
The non-crystalline nature of 6b presumably arises due to chain disorder from linear 1,3-repulsions between the fluorines, so the preferred conformation of this motif could not be determined in this study. The melting point of palmitic acid 6c (89.9 °C) was notable in that it was significantly higher than that of the two other analogues 6a and 6b, and also of palmitic acid itself. The solid state structure of 6c and 27 show that the 1,4-di-CF2 motif prefers an anti-zig-zag conformation. We attribute this preference to intramolecular dipole–dipole relaxation which is maximised in the extended anti-zig-zag chain conformation (Figure 8). Also repulsive through space 1,4-F···F interactions will be disfavoured if the chain undergoes gauche conformational disorder. These contributing factors suggest that the 1,4-di-CF2 motif (R–CF2CH2CH2CF2–R) will be useful for adding conformational stability to aliphatic chains.
![[1860-5397-10-4-8]](/bjoc/content/figures/1860-5397-10-4-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Conformational interconversion of 1,4-di-CF2 motif.
Figure 8: Conformational interconversion of 1,4-di-CF2 motif.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 4.9 MB | Download |
References
-
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Liu, W.; Huang, W.; Cheng, M.-J.; Nielson, R. J.; Goddard, W. A., III; Groves, J. T. Science 2012, 337, 1322–1325. doi:10.1126/science.1222327
Return to citation in text: [1] -
Jarchow-Choy, S. K.; Sjuvarsson, E.; Sintim, H. O.; Eriksson, S.; Kool, E. T. J. Am. Chem. Soc. 2009, 131, 5488–5494. doi:10.1021/ja808244t
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060
Return to citation in text: [1] [2] [3] -
O’Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26
Return to citation in text: [1] [2] -
Urbina-Blanco, C. A.; Skibiński, M.; O'Hagan, D.; Nolan, S. P. Chem. Commun. 2013, 49, 7201–7203. doi:10.1039/c3cc44312d
Return to citation in text: [1] [2] -
Dasaradhi, L.; O'Hagan, D.; Petty, M. C.; Pearson, C. J. Chem. Soc., Perkin Trans. 2 1995, 221–225. doi:10.1039/p29950000221
Return to citation in text: [1] -
Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045
Return to citation in text: [1] [2] -
O’ Hagan, D. J. Fluorine Chem. 1989, 43, 371–377. doi:10.1016/S0022-1139(00)82723-2
Return to citation in text: [1] [2] -
Middleton, W. J. J. Org. Chem. 1975, 40, 574–578. doi:10.1021/jo00893a007
Return to citation in text: [1] -
O'Hagan, D.; Al-Maharik, N. Aldrichimica Acta 2011, 44, 65–75.
Return to citation in text: [1] -
Prakesch, M.; Kerouredan, E.; Grée, D.; Grée, R.; DeChancie, J.; Houk, K. N. J. Fluorine Chem. 2004, 125, 537–541. doi:10.1016/j.jfluchem.2003.11.027
Return to citation in text: [1] [2] -
Khalaf, A.; Grée, D.; Abdallah, H.; Jaber, N.; Hachem, A.; Grée, R. Tetrahedron 2011, 67, 3881–3886. doi:10.1016/j.tet.2011.03.073
Return to citation in text: [1] [2] -
Bannwarth, P.; Valleix, A.; Grée, D.; Grée, R. J. Org. Chem. 2009, 74, 4646–4649. doi:10.1021/jo900674u
Return to citation in text: [1] [2] -
Bannwarth, P.; Grée, D.; Grée, R. Tetrahedron Lett. 2010, 51, 2413–2415. doi:10.1016/j.tetlet.2010.02.116
Return to citation in text: [1] [2] -
Pacheco, M. C.; Purser, S.; Gouverneur, V. Chem. Rev. 2008, 108, 1943–1981. doi:10.1021/cr068410e
Return to citation in text: [1] -
Pajkert, R.; Röschenthaler, G.-V. J. Org. Chem. 2013, 78, 3697–3708. doi:10.1021/jo400198a
Return to citation in text: [1] -
Drakesmith, F. G.; Stewart, O. J.; Tarrant, P. J. Org. Chem. 1968, 33, 280–285. doi:10.1021/jo01265a055
Return to citation in text: [1] -
Howard, J. A. K.; Hoy, V. J.; O’Hagan, D.; Smith, G. T. Tetrahedron 1996, 52, 12613–12622. doi:10.1016/0040-4020(96)00749-1
Return to citation in text: [1]
| 1. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 2. | Liu, W.; Huang, W.; Cheng, M.-J.; Nielson, R. J.; Goddard, W. A., III; Groves, J. T. Science 2012, 337, 1322–1325. doi:10.1126/science.1222327 |
| 3. | Jarchow-Choy, S. K.; Sjuvarsson, E.; Sintim, H. O.; Eriksson, S.; Kool, E. T. J. Am. Chem. Soc. 2009, 131, 5488–5494. doi:10.1021/ja808244t |
| 4. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 8. | Urbina-Blanco, C. A.; Skibiński, M.; O'Hagan, D.; Nolan, S. P. Chem. Commun. 2013, 49, 7201–7203. doi:10.1039/c3cc44312d |
| 21. | Howard, J. A. K.; Hoy, V. J.; O’Hagan, D.; Smith, G. T. Tetrahedron 1996, 52, 12613–12622. doi:10.1016/0040-4020(96)00749-1 |
| 8. | Urbina-Blanco, C. A.; Skibiński, M.; O'Hagan, D.; Nolan, S. P. Chem. Commun. 2013, 49, 7201–7203. doi:10.1039/c3cc44312d |
| 6. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 7. | O’Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26 |
| 14. | Prakesch, M.; Kerouredan, E.; Grée, D.; Grée, R.; DeChancie, J.; Houk, K. N. J. Fluorine Chem. 2004, 125, 537–541. doi:10.1016/j.jfluchem.2003.11.027 |
| 15. | Khalaf, A.; Grée, D.; Abdallah, H.; Jaber, N.; Hachem, A.; Grée, R. Tetrahedron 2011, 67, 3881–3886. doi:10.1016/j.tet.2011.03.073 |
| 16. | Bannwarth, P.; Valleix, A.; Grée, D.; Grée, R. J. Org. Chem. 2009, 74, 4646–4649. doi:10.1021/jo900674u |
| 17. | Bannwarth, P.; Grée, D.; Grée, R. Tetrahedron Lett. 2010, 51, 2413–2415. doi:10.1016/j.tetlet.2010.02.116 |
| 19. | Pajkert, R.; Röschenthaler, G.-V. J. Org. Chem. 2013, 78, 3697–3708. doi:10.1021/jo400198a |
| 20. | Drakesmith, F. G.; Stewart, O. J.; Tarrant, P. J. Org. Chem. 1968, 33, 280–285. doi:10.1021/jo01265a055 |
| 10. | Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045 |
| 11. | O’ Hagan, D. J. Fluorine Chem. 1989, 43, 371–377. doi:10.1016/S0022-1139(00)82723-2 |
| 14. | Prakesch, M.; Kerouredan, E.; Grée, D.; Grée, R.; DeChancie, J.; Houk, K. N. J. Fluorine Chem. 2004, 125, 537–541. doi:10.1016/j.jfluchem.2003.11.027 |
| 15. | Khalaf, A.; Grée, D.; Abdallah, H.; Jaber, N.; Hachem, A.; Grée, R. Tetrahedron 2011, 67, 3881–3886. doi:10.1016/j.tet.2011.03.073 |
| 16. | Bannwarth, P.; Valleix, A.; Grée, D.; Grée, R. J. Org. Chem. 2009, 74, 4646–4649. doi:10.1021/jo900674u |
| 17. | Bannwarth, P.; Grée, D.; Grée, R. Tetrahedron Lett. 2010, 51, 2413–2415. doi:10.1016/j.tetlet.2010.02.116 |
| 18. | Pacheco, M. C.; Purser, S.; Gouverneur, V. Chem. Rev. 2008, 108, 1943–1981. doi:10.1021/cr068410e |
| 9. | Dasaradhi, L.; O'Hagan, D.; Petty, M. C.; Pearson, C. J. Chem. Soc., Perkin Trans. 2 1995, 221–225. doi:10.1039/p29950000221 |
| 10. | Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045 |
| 11. | O’ Hagan, D. J. Fluorine Chem. 1989, 43, 371–377. doi:10.1016/S0022-1139(00)82723-2 |
| 6. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 7. | O’Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26 |
| 6. | Skibinski, M.; Wang, Y.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Angew. Chem., Int. Ed. 2011, 50, 10581–10584. doi:10.1002/anie.201105060 |
| 12. | Middleton, W. J. J. Org. Chem. 1975, 40, 574–578. doi:10.1021/jo00893a007 |
| 13. | O'Hagan, D.; Al-Maharik, N. Aldrichimica Acta 2011, 44, 65–75. |
© 2014 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)