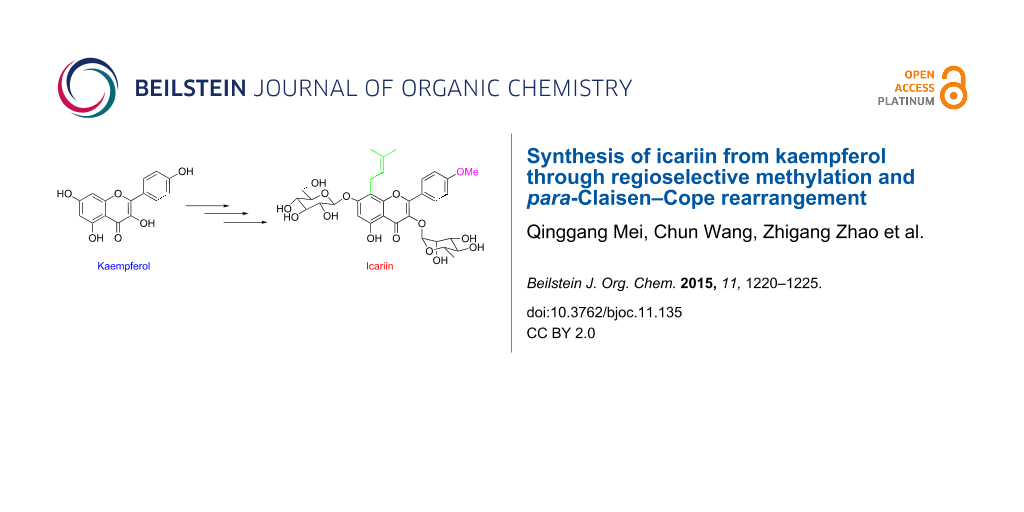
Abstract
The hemisynthesis of the naturally occurring bioactive flavonoid glycoside icariin (1) has been accomplished in eleven steps with 7% overall yield from kaempferol. The 4′-OH methylation of kaempferol, the 8-prenylation of 3-O-methoxymethyl-4′-O-methyl-5-O-prenyl-7-O-benzylkaempferol (8) via para-Claisen–Cope rearrangement catalyzed by Eu(fod)3 in the presence of NaHCO3, and the glycosylation of icaritin (3) are the key steps.

Graphical Abstract
Introduction
The plants of the Genus Epimedium, also known as “Yin-Yang-Huo”, were used in traditional Chinese medicine and are believed to invigorate the kidney and enhance the “Yang”. For more than one thousand years, some plants of Epimedium have been widely used in China to treat cardiovascular diseases, amnesia, arthritis, asthenia, impotence, infertility, lumbago and other chronic illnesses [1,2]. Icariin (1), a 3,7-diglycosylflavone (Figure 1), is recognized as the major pharmacologically active ingredient of these plants [3,4], and has been used as the index for quality control of the herbs and relative drug preparations [5]. Icariin, icariside I (2), and their aglycone, icaritin (3) (Figure 1), possess multiple biological activities, such as anti-oxidative [6], anti-inflammatory [7], anti-osteoporotic [8], anticancer [9], neuroprotective [10], angiogenesis stimulating [11], testosterone mimetic [12], antidepressant-like [13] and multidrug resistance reversal [14] activities.
![[1860-5397-11-135-1]](/bjoc/content/figures/1860-5397-11-135-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of icariin (1), icariside I (2) and icaritin (3).
Figure 1: Structures of icariin (1), icariside I (2) and icaritin (3).
Although icariin exerts a variety of important bioactivities, reports concerning its synthesis are very scarce. One Chinese patent reported a 15-step total synthesis of icariin from benzyl alcohol with 0.6% overall yield [15]. The 11-step synthesis of icaritin, starting from 2,4,6-trihydroxyacetophenone via microwave-assisted Claisen rearrangement reaction as the key step, was succeeded with an overall yield of 23% [16]. In view of the long synthetic routes, tedious work-up and harsh reaction conditions, an alternative access to icariin (1) and related compounds is demanded for large-scale production and practical application.
Our interest in flavonoids with activities such as estrogen biosynthesis regulation and PDE inhibition, prompted us to seek for a practically synthetic approach to icariin. Previously our group prepared icaritin from phloroglucin through an Algar–Flynn–Oyamada reaction and europium-promoted prenylation [17]. However, a number of challengable problems such as strict conditions, numerous byproducts and poor yields, were calling for our continuous efforts to overcome these hitches [18]. As a continuation of this program, herein we report a new approach to icaritin and then icariin through semi-synthesis from the commercially available natural product kaempferol. Our previously developed regioselective methylation of kaempferol [19], Europium(III)-catalyzed para-Claisen–Cope rearrangement and the bis-glycosylation are the key features of this linear synthesis. Previously, we succeeded in the selective methylation of 4′-OH in kaempferol. In this work, we focus on developing an efficient procedure for the selective prenylation of flavonols for facile access to icariin (1).
Results and Discussion
Our synthetic approach to 1 commenced with the preparation of aglycone 3, as illustrated in Scheme 1. First, 7-O-benzylkaempferide (6) was easily obtained from kaempferol through tetraacetylation, followed by benzylation at C-7 and selectively methylation, according to our previously developed procedure [19]. In order to methylate exclusively the 4′-OH in kaempferol, we initially attempted to use methoxymethyl (MOM) as 7-OH protecting group, but this method could not provide an ideal yield in the 4′-OH selective methylation and in subsequent Claisen–Cope rearrangement.
![[1860-5397-11-135-i1]](/bjoc/content/inline/1860-5397-11-135-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (a) Ac2O, pyridine, 94%; (b) BnBr, KI, K2CO3, acetone, 85%; (c) Me2SO4, K2CO3, acetone, MeOH, 82%; (d) MOMCl, N,N-diisopropylethylamine (DIPEA), CH2Cl2, 93%; (e) 3,3-dimethylallyl bromide, 18-crown-6, K2CO3, acetone, 86%; (f) Eu(fod)3, NaHCO3, PhCl, 85 °C, 61%; (g) MeOH, 3 M HCl (aq), reflux, 95%; (h) Pd/C, 1,4-cyclohexadiene, MeOH, 84%.
Scheme 1: Reagents and conditions: (a) Ac2O, pyridine, 94%; (b) BnBr, KI, K2CO3, acetone, 85%; (c) Me2SO4, K2...
The resulting free 3-OH in 6 is protected with an orthogonal protecting group to give 3-O-methoxymethyl-4′-O-methyl-7-O-benzylkaempferol (7) in 93% yield. Then introduction of a requisite prenyl ether moiety at C-5 of 7, with 3,3-dimethylallyl bromide in the presence of potassium carbonate and 18-crown-6, formed 8, whose all hydroxy groups were protected for the rearrangement accompanied in the next step. It is noticeable that this compound is slightly less stable in solution, easily decomposing into 7 and 11 (Scheme 2), especially under acidic conditions.
![[1860-5397-11-135-i2]](/bjoc/content/inline/1860-5397-11-135-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The Claisen rearrangement is commonly accepted as an efficient method for the preparation of C-isopentenyl. In the case of flavonoids, control of the regioselectivity of ortho (C6)/para (C8)-rearranged products is still remained a challenging issue [20]. Once the prenyl ether 8 was obtained, attention was fastened on the para-Claisen–Cope rearrangement to acquire the 8-prenylated product. Two pathways have been developed for the rearrangement: direct heating at about 217 °C [17,21] and heating at about 60 °C in the presence of Eu(fod)3 [22,23]. When we adapted the former path, reflux of 8 in N,N-diethylaniline for 3 h led to complete consumption of 8 and gave an intractable product mixture, probably due to the prevalence of the 5-O-prenyl chain elimination with no rearrangement. Then we turned to try the latter method with 10 mol % Eu(fod)3 as the catalyst at 60 °C in dry CHCl3 (Table 1), in which the Claisen rearrangement of 8 indeed took place. Unlike reported results [24-26], we obtained the ortho-rearranged product, which was not the 6-(1,1-dimethylallyl) product 12a, but an inseparable epimeric mixture of the dihydrofuro-flavonol 12, presumably resulted from the cyclization of the 6-(1,1-dimethylallyl) chain with the 5-hydroxy group (Scheme 3). The ratio of the para-rearranged product 9 to 12 was 0.7:1. When PhCl was used as solvent, the rearrangement of 8 at 85 ºC for 24 h allowed the isolation of 9 and 12 in a 0.9:1 ratio (Table 1). Although the proportion of 9 and the total yield were improved, the para/ortho-selectivity was still unsatisfactory, even when prolonging the reaction time to 36 h or raising the temperature to 100 °C. By carefully investigating the two routes in Scheme 3, we assumed that an appropriate base is in favor of the [1,5]H σ migration in route 2, as well as lowering the acidity of Eu(fod)3 to prevent 12b from cyclization in route 1, and therefore enhances the proportion of the para-rearrangement product 8. Obviously, strong alkali could lead to heterocycle cleavage of the flavonoid skeleton. Gratifyingly, when prenyl ether 8 was treated with catalytic amounts of Eu(fod)3 and NaHCO3 in PhCl at 85 °C for 24 h (Table 1), the Claisen para-rearranged product was obtained selectively in 61% yield with the ratio 9 to 12 of 2.1:1. With other bases such as K2CO3 or DIPEA, the results were very different, probably due to the partial inactivity of Eu(fod)3 under these conditions. It is noteworthy that NaHCO3 only affected the 9/12 ratio, but not the total amount of C-isoprenoid flavonols 9 and 12, which was found to stay around 90%. This to some extent, implies that our hypothesis on using appropriate base to hinder the Route 1 and to promote the Cope rearrangement step from C-6 to C-8 could be true. Therefore, the combination of Eu(fod)3 and NaHCO3 is preferable for the Claisen–Cope rearrangement from 5-O- to 8-C-prenyl products.
Table 1: Rearrangement of 5-O-prenylflavone 8.
| Methoda | T (ºC) | t (h) | Products (%)b | Ratio 9:12 | Total 9 + 12 |
|---|---|---|---|---|---|
| CHCl3, Eu(fod)3 | 60 | 24 | 9 (33), 12 (46) | 0.7:1 | 79% |
| PhCl, Eu(fod)3 | 85 | 24 | 9 (42), 12 (47) | 0.9:1 | 89% |
| PhCl, Eu(fod)3, NaHCO3 | 85 | 24 | 9 (61), 12 (29) | 2.1:1 | 90% |
aEu(fod)3: 10 mol %; NaHCO3: 100 mol %. bIsolated yield.
![[1860-5397-11-135-i3]](/bjoc/content/inline/1860-5397-11-135-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Claisen rearrangement of flavonol 8.
Scheme 3: Claisen rearrangement of flavonol 8.
Exposure of compound 9 to dilute methanolic HCl at reflux resulted in the cleavage of the methoxymethyl group, giving rise to 7-O-benzylicaritin (10). Removal of the benzyl group in 10 resulting in 3 without affecting the double bond of the prenyl residue was another challenge. The hydrogenation of 10 with H2–Pd/C using EtOAc/MeOH (1:1) as solvent at 10 °C for 3 h, followed by column chromatography and recrystallization, provided 3 in 39% yield along with a large amount of the byproduct with reduced double bond. Then we tried a transfer hydrogenolysis method using ammonium formate/10% Pd/C [27], but the result was not improved. The desired product icaritin (3) was finally obtained in 84% yield from 10 when we used 1,4-cyclohexadiene/10% Pd/C in MeOH [28]. Hence, we optimized a convenient eight-step sequence from kaempferol to 3 through regioselective methylation and a domino Claisen–Cope rearrangement that delivered 3 in 26% overall yield. The NMR spectra of synthetic 3 (see Supporting Information File 1 for experimental and spectral data) were in accordance with the reported ones [16,29].
With icaritin (3) in hand, the selective glycosylation was investigated (Scheme 4). The alkylation of OH in kaempferol followed a specific reactivity order: 7 > 4′ > 3 >> 5 [19]. We initially attempted the 7-OH glycosylation with tetra-O-acetylglucopyranosyl bromide (15) [30] as the donor and SrCO3 or AgNO3 as an activator in pyridine or quinoline [15,31], but it was unsuccessful. TLC monitoring revealed that most of icartin was still unreacted even prolonging the reaction time to 48 h. To our delight, switching the solvent to DMF/CHCl3 and the activator to Ag2CO3 made the reaction to proceed smoothly, resulting in the synthesis of 13. The yellow powder 13 was treated with tri-O-acetylrhamnopyranosyl bromide (16) [30] in CH2Cl2 using Ag2O as activator, following a known procedure [32]. Compound 14 was easily separated by normal silica gel column chromatography, with a yield of 31% from 3.
![[1860-5397-11-135-i4]](/bjoc/content/inline/1860-5397-11-135-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: (a) 15, DMF/CHCl3, Ag2CO3, molecular sieves (4 Å, powder); (b) 16, CH2Cl2, Ag2O, molecular sieves (4 Å powder), 31% for 2 steps; (c) NH3 (g), MeOH, 94%; (d) NH3 (g), MeOH, 63% for 2 steps.
Scheme 4: Reagents and conditions: (a) 15, DMF/CHCl3, Ag2CO3, molecular sieves (4 Å, powder); (b) 16, CH2Cl2,...
The final procedure in completing the synthesis of icariin (1) is the removal of all acetyl groups in 14 (Scheme 4). In contrast to the traditional method for deacetylation under basic conditions such as with CH3ONa [15] or K2CO3 [33] in CH3OH, employing methanolic ammonia (7.0 M) for 3 h at room temperature effectively suppressed the cleavage of the sugar moiety [34] and gave the target compound 1 in high conversion. Icariside I (2) was also obtained using the same procedure from compound 13. Thus, icariin (1) and icariside I (2) were synthesized from kaempferol with 7% and 16% overall yields, respectively.
The compounds 14, 2 and 1 were fully characterized by ESI-HRMS, 1H NMR, 13C NMR, 1H,1H COSY, 1H,13C HMBC/HSQC and IR (see Supporting Information File 1 for experimental and spectral data). In icariin (1), the configurations of the anomeric C-atoms of rhamnose and glocuse were α and β on the basis of the anomeric proton signals at δ 5.27 and 5.00 with J = 1.3 Hz and 7.4 Hz, respectively. The NMR data of icariin (1) were in agreement with those reported [29,35] and with the spectra of the authentic, naturally derived sample.
Conclusion
In conclusion, we have developed an efficient and practical procedure for the preparation of the pharmacologically important compounds icariin (1) and icariside I (2) from kaempferol with 7% and 16% overall yields, respectively. The addition of NaHCO3 to the Eu(fod)3-catalyzed Claisen–Cope rearrangement could inhibit the formation of the ortho-cyclized rearrangement product, thus selectively promoting the para-Claisen–Cope rearrangement of flavonoid 5-prenyl ether. This procedure could afford facile access to the derivatives of icariin (1) and other structurally related flavonoid analogues.
Supporting Information
| Supporting Information File 1: Experimental section and copies of NMR, ESI-HRMS, HMBC, HSQC, COSY and NOESY spectra. | ||
| Format: PDF | Size: 9.0 MB | Download |
References
-
Ma, H.; He, X.; Yang, Y.; Li, M.; Hao, D.; Jia, Z. J. Ethnopharmacol. 2011, 134, 519–541. doi:10.1016/j.jep.2011.01.001
Return to citation in text: [1] -
Edouard, M. J.; Miao, L.; Fan, G.-W.; Ojong, B. B. O.; Zhen, H.; Zhang, J.; Gao, X.-M.; Zhu, Y. Chin. J. Nat. Med. 2014, 12, 321–334. doi:10.1016/S1875-5364(14)60040-6
Return to citation in text: [1] -
Yang, L.; Lu, D.; Guo, J.; Meng, X.; Zhang, G.; Wang, F. J. Ethnopharmacol. 2013, 145, 715–721. doi:10.1016/j.jep.2012.11.031
Return to citation in text: [1] -
Huang, J.; Yuan, L.; Wang, X.; Zhang, T.-L.; Wang, K. Life Sci. 2007, 81, 832–840. doi:10.1016/j.lfs.2007.07.015
Return to citation in text: [1] -
Chinese Pharmacopoeia Commission. Herba Epimedii. Chinese Pharmacopoeia, Part I; Chinese Pharmacopoeia Commission; China Medical Science Press: Beijing, 2010; pp 306–307.
Return to citation in text: [1] -
Liu, P.; Jin, X.; Lv, H.; Li, J.; Xu, W.; Qian, H.; Yin, Z. In Vitro Cell. Dev. Biol.: Anim. 2014, 50, 899–908. doi:10.1007/s11626-014-9792-8
Return to citation in text: [1] -
Xu, C.-Q.; Liu, B.-J.; Wu, J.-F.; Xu, Y.-C.; Duan, X.-H.; Cao, Y.-X.; Dong, J.-C. Eur. J. Pharmacol. 2010, 642, 146–153. doi:10.1016/j.ejphar.2010.05.012
Return to citation in text: [1] -
Zhai, Y.-K.; Guo, X.-Y.; Ge, B.-F.; Zhen, P.; Ma, X.-N.; Zhou, J.; Ma, H.-P.; Xian, C. J.; Chen, K.-M. Bone 2014, 66, 189–198. doi:10.1016/j.bone.2014.06.016
Return to citation in text: [1] -
Wu, T.; Wang, S.; Wu, J.; Lin, Z.; Sui, X.; Xu, X.; Shimizu, N.; Chen, B.; Wang, X. J. Exp. Clin. Cancer Res. 2015, 34, No. 17. doi:10.1186/s13046-015-0133-x
Return to citation in text: [1] -
Wang, X.; Li, J.; Qian, L.; Zang, X.-F.; Zhang, S.-Y.; Wang, X.-Y.; Jin, J.-L.; Zhu, X.-L.; Zhang, X.-B.; Wang, Z.-Y.; Xu, Y. Neuroscience 2013, 236, 281–288. doi:10.1016/j.neuroscience.2012.12.074
Return to citation in text: [1] -
Chung, B.-H.; Kim, J.-D.; Kim, C.-K.; Kim, J. H.; Won, M.-H.; Lee, H.-S.; Dong, M.-S.; Ha, K.-S.; Kwon, Y.-G.; Kim, Y.-M. Biochem. Biophys. Res. Commun. 2008, 376, 404–408. doi:10.1016/j.bbrc.2008.09.001
Return to citation in text: [1] -
Shindel, A. W.; Xin, Z.-C.; Lin, G.; Fandel, T. M.; Huang, Y.-C.; Banie, L.; Breyer, B. N.; Garcia, M. M.; Lin, C.-S.; Lue, T. F. J. Sex. Med. 2010, 7, 1518–1528. doi:10.1111/j.1743-6109.2009.01699.x
Return to citation in text: [1] -
Wu, J.; Du, J.; Xu, C.; Le, J.; Xu, Y.; Liu, B.; Dong, J. Pharmacol., Biochem. Behav. 2011, 98, 273–278. doi:10.1016/j.pbb.2011.01.008
Return to citation in text: [1] -
Sun, L.; Chen, W.; Qu, L.; Wu, J.; Si, J. Mol. Med. Rep. 2013, 8, 1883–1887. doi:10.3892/mmr.2013.1742
Return to citation in text: [1] -
Li, Y.-L.; Zhu, Y.-C.; Zhang, J.; Cui, L.-Y. Total synthesis method of natural product barrenwort glycosides compounds. Chinese Patent CN101,205,223, June 25, 2008.
Return to citation in text: [1] [2] [3] -
Nguyen, V.-S.; Shi, L.; Li, Y.; Wang, Q.-A. Lett. Org. Chem. 2014, 11, 677–681. doi:10.2174/157017861109140903103927
Return to citation in text: [1] [2] -
Mu, G.; Pu, W.; Zhou, M.; Liu, Y.; Yang, H.; Wang, C. Chin. J. Org. Chem. 2013, 33, 1298–1303. doi:10.6023/cjoc201303016
Return to citation in text: [1] [2] -
Mei, Q.; Yuan, W.; Wang, C. Chin. J. Org. Chem. 2015, 35, 70–84. doi:10.6023/cjoc201408038
Return to citation in text: [1] -
Mei, Q.; Wang, C.; Yuan, W.; Zhang, G. Beilstein J. Org. Chem. 2015, 11, 288–293. doi:10.3762/bjoc.11.33
Return to citation in text: [1] [2] [3] -
Tischer, S.; Metz, P. Adv. Synth. Catal. 2007, 349, 147–151. doi:10.1002/adsc.200600454
Return to citation in text: [1] -
Daskiewicz, J.-B.; Depeint, F.; Viornery, L.; Bayet, C.; Comte-Sarrazin, G.; Comte, G.; Gee, J. M.; Johnson, I. T.; Ndjoko, K.; Hostettmann, K.; Barron, D. J. Med. Chem. 2005, 48, 2790–2804. doi:10.1021/jm040770b
Return to citation in text: [1] -
Al-Maharik, N.; Botting, N. P. Tetrahedron 2003, 59, 4177–4181. doi:10.1016/S0040-4020(03)00579-9
Return to citation in text: [1] -
Poerwono, H.; Sasaki, S.; Hattori, Y.; Higashiyama, K. Bioorg. Med. Chem. Lett. 2010, 20, 2086–2089. doi:10.1016/j.bmcl.2010.02.068
Return to citation in text: [1] -
Daskiewicz, J.-B.; Bayet, C.; Barron, D. Tetrahedron 2002, 58, 3589–3595. doi:10.1016/S0040-4020(02)00309-5
Return to citation in text: [1] -
Gester, S.; Metz, P.; Zierau, O.; Vollmer, G. Tetrahedron 2001, 57, 1015–1018. doi:10.1016/S0040-4020(00)01078-4
Return to citation in text: [1] -
Daskiewicz, J.-B.; Bayet, C.; Barron, D. Tetrahedron Lett. 2001, 42, 7241–7244. doi:10.1016/S0040-4039(01)01503-9
Return to citation in text: [1] -
Lohray, B. B.; Jayamma, Y.; Chatterjee, M. J. Org. Chem. 1995, 60, 5958–5960. doi:10.1021/jo00123a037
Return to citation in text: [1] -
Knapp, S.; Nandan, S. R. J. Org. Chem. 1994, 59, 281–283. doi:10.1021/jo00081a004
Return to citation in text: [1] -
Dell’Agli, M.; Galli, G. V.; Dal Cero, E.; Belluti, F.; Matera, R.; Zironi, E.; Pagliuca, G.; Bosisio, E. J. Nat. Prod. 2008, 71, 1513–1517. doi:10.1021/np800049y
Return to citation in text: [1] [2] -
Li, Y.-W.; Li, Y.-X.; Zhang, W.; Guan, H.-S. Chin. J. Org. Chem. 2004, 24, 438–439.
Return to citation in text: [1] [2] -
Wang, B.-B.; Zhang, S. Method for synthesizing icariin by glucosidation of dEpimedium herb. Chinese Patent CN102,093,450, June 15, 2011.
Return to citation in text: [1] -
Maloney, D. J.; Hecht, S. M. Org. Lett. 2005, 7, 1097–1099. doi:10.1021/ol0500463
Return to citation in text: [1] -
Urgaonkar, S.; Shaw, J. T. J. Org. Chem. 2007, 72, 4582–4585. doi:10.1021/jo070502w
Return to citation in text: [1] -
Yang, X.; Sun, Y.; Xu, Q.; Guo, Z. Org. Biomol. Chem. 2006, 4, 2483–2491. doi:10.1039/b604521a
Return to citation in text: [1] -
Liu, R.; Li, A.; Sun, A.; Cui, J.; Kong, L. J. Chromatogr. A 2005, 1064, 53–57. doi:10.1016/j.chroma.2004.12.026
Return to citation in text: [1]
| 15. | Li, Y.-L.; Zhu, Y.-C.; Zhang, J.; Cui, L.-Y. Total synthesis method of natural product barrenwort glycosides compounds. Chinese Patent CN101,205,223, June 25, 2008. |
| 31. | Wang, B.-B.; Zhang, S. Method for synthesizing icariin by glucosidation of dEpimedium herb. Chinese Patent CN102,093,450, June 15, 2011. |
| 30. | Li, Y.-W.; Li, Y.-X.; Zhang, W.; Guan, H.-S. Chin. J. Org. Chem. 2004, 24, 438–439. |
| 32. | Maloney, D. J.; Hecht, S. M. Org. Lett. 2005, 7, 1097–1099. doi:10.1021/ol0500463 |
| 1. | Ma, H.; He, X.; Yang, Y.; Li, M.; Hao, D.; Jia, Z. J. Ethnopharmacol. 2011, 134, 519–541. doi:10.1016/j.jep.2011.01.001 |
| 2. | Edouard, M. J.; Miao, L.; Fan, G.-W.; Ojong, B. B. O.; Zhen, H.; Zhang, J.; Gao, X.-M.; Zhu, Y. Chin. J. Nat. Med. 2014, 12, 321–334. doi:10.1016/S1875-5364(14)60040-6 |
| 7. | Xu, C.-Q.; Liu, B.-J.; Wu, J.-F.; Xu, Y.-C.; Duan, X.-H.; Cao, Y.-X.; Dong, J.-C. Eur. J. Pharmacol. 2010, 642, 146–153. doi:10.1016/j.ejphar.2010.05.012 |
| 17. | Mu, G.; Pu, W.; Zhou, M.; Liu, Y.; Yang, H.; Wang, C. Chin. J. Org. Chem. 2013, 33, 1298–1303. doi:10.6023/cjoc201303016 |
| 6. | Liu, P.; Jin, X.; Lv, H.; Li, J.; Xu, W.; Qian, H.; Yin, Z. In Vitro Cell. Dev. Biol.: Anim. 2014, 50, 899–908. doi:10.1007/s11626-014-9792-8 |
| 18. | Mei, Q.; Yuan, W.; Wang, C. Chin. J. Org. Chem. 2015, 35, 70–84. doi:10.6023/cjoc201408038 |
| 5. | Chinese Pharmacopoeia Commission. Herba Epimedii. Chinese Pharmacopoeia, Part I; Chinese Pharmacopoeia Commission; China Medical Science Press: Beijing, 2010; pp 306–307. |
| 15. | Li, Y.-L.; Zhu, Y.-C.; Zhang, J.; Cui, L.-Y. Total synthesis method of natural product barrenwort glycosides compounds. Chinese Patent CN101,205,223, June 25, 2008. |
| 3. | Yang, L.; Lu, D.; Guo, J.; Meng, X.; Zhang, G.; Wang, F. J. Ethnopharmacol. 2013, 145, 715–721. doi:10.1016/j.jep.2012.11.031 |
| 4. | Huang, J.; Yuan, L.; Wang, X.; Zhang, T.-L.; Wang, K. Life Sci. 2007, 81, 832–840. doi:10.1016/j.lfs.2007.07.015 |
| 16. | Nguyen, V.-S.; Shi, L.; Li, Y.; Wang, Q.-A. Lett. Org. Chem. 2014, 11, 677–681. doi:10.2174/157017861109140903103927 |
| 11. | Chung, B.-H.; Kim, J.-D.; Kim, C.-K.; Kim, J. H.; Won, M.-H.; Lee, H.-S.; Dong, M.-S.; Ha, K.-S.; Kwon, Y.-G.; Kim, Y.-M. Biochem. Biophys. Res. Commun. 2008, 376, 404–408. doi:10.1016/j.bbrc.2008.09.001 |
| 13. | Wu, J.; Du, J.; Xu, C.; Le, J.; Xu, Y.; Liu, B.; Dong, J. Pharmacol., Biochem. Behav. 2011, 98, 273–278. doi:10.1016/j.pbb.2011.01.008 |
| 34. | Yang, X.; Sun, Y.; Xu, Q.; Guo, Z. Org. Biomol. Chem. 2006, 4, 2483–2491. doi:10.1039/b604521a |
| 10. | Wang, X.; Li, J.; Qian, L.; Zang, X.-F.; Zhang, S.-Y.; Wang, X.-Y.; Jin, J.-L.; Zhu, X.-L.; Zhang, X.-B.; Wang, Z.-Y.; Xu, Y. Neuroscience 2013, 236, 281–288. doi:10.1016/j.neuroscience.2012.12.074 |
| 14. | Sun, L.; Chen, W.; Qu, L.; Wu, J.; Si, J. Mol. Med. Rep. 2013, 8, 1883–1887. doi:10.3892/mmr.2013.1742 |
| 29. | Dell’Agli, M.; Galli, G. V.; Dal Cero, E.; Belluti, F.; Matera, R.; Zironi, E.; Pagliuca, G.; Bosisio, E. J. Nat. Prod. 2008, 71, 1513–1517. doi:10.1021/np800049y |
| 35. | Liu, R.; Li, A.; Sun, A.; Cui, J.; Kong, L. J. Chromatogr. A 2005, 1064, 53–57. doi:10.1016/j.chroma.2004.12.026 |
| 9. | Wu, T.; Wang, S.; Wu, J.; Lin, Z.; Sui, X.; Xu, X.; Shimizu, N.; Chen, B.; Wang, X. J. Exp. Clin. Cancer Res. 2015, 34, No. 17. doi:10.1186/s13046-015-0133-x |
| 15. | Li, Y.-L.; Zhu, Y.-C.; Zhang, J.; Cui, L.-Y. Total synthesis method of natural product barrenwort glycosides compounds. Chinese Patent CN101,205,223, June 25, 2008. |
| 8. | Zhai, Y.-K.; Guo, X.-Y.; Ge, B.-F.; Zhen, P.; Ma, X.-N.; Zhou, J.; Ma, H.-P.; Xian, C. J.; Chen, K.-M. Bone 2014, 66, 189–198. doi:10.1016/j.bone.2014.06.016 |
| 12. | Shindel, A. W.; Xin, Z.-C.; Lin, G.; Fandel, T. M.; Huang, Y.-C.; Banie, L.; Breyer, B. N.; Garcia, M. M.; Lin, C.-S.; Lue, T. F. J. Sex. Med. 2010, 7, 1518–1528. doi:10.1111/j.1743-6109.2009.01699.x |
| 33. | Urgaonkar, S.; Shaw, J. T. J. Org. Chem. 2007, 72, 4582–4585. doi:10.1021/jo070502w |
| 20. | Tischer, S.; Metz, P. Adv. Synth. Catal. 2007, 349, 147–151. doi:10.1002/adsc.200600454 |
| 19. | Mei, Q.; Wang, C.; Yuan, W.; Zhang, G. Beilstein J. Org. Chem. 2015, 11, 288–293. doi:10.3762/bjoc.11.33 |
| 19. | Mei, Q.; Wang, C.; Yuan, W.; Zhang, G. Beilstein J. Org. Chem. 2015, 11, 288–293. doi:10.3762/bjoc.11.33 |
| 19. | Mei, Q.; Wang, C.; Yuan, W.; Zhang, G. Beilstein J. Org. Chem. 2015, 11, 288–293. doi:10.3762/bjoc.11.33 |
| 30. | Li, Y.-W.; Li, Y.-X.; Zhang, W.; Guan, H.-S. Chin. J. Org. Chem. 2004, 24, 438–439. |
| 28. | Knapp, S.; Nandan, S. R. J. Org. Chem. 1994, 59, 281–283. doi:10.1021/jo00081a004 |
| 16. | Nguyen, V.-S.; Shi, L.; Li, Y.; Wang, Q.-A. Lett. Org. Chem. 2014, 11, 677–681. doi:10.2174/157017861109140903103927 |
| 29. | Dell’Agli, M.; Galli, G. V.; Dal Cero, E.; Belluti, F.; Matera, R.; Zironi, E.; Pagliuca, G.; Bosisio, E. J. Nat. Prod. 2008, 71, 1513–1517. doi:10.1021/np800049y |
| 24. | Daskiewicz, J.-B.; Bayet, C.; Barron, D. Tetrahedron 2002, 58, 3589–3595. doi:10.1016/S0040-4020(02)00309-5 |
| 25. | Gester, S.; Metz, P.; Zierau, O.; Vollmer, G. Tetrahedron 2001, 57, 1015–1018. doi:10.1016/S0040-4020(00)01078-4 |
| 26. | Daskiewicz, J.-B.; Bayet, C.; Barron, D. Tetrahedron Lett. 2001, 42, 7241–7244. doi:10.1016/S0040-4039(01)01503-9 |
| 27. | Lohray, B. B.; Jayamma, Y.; Chatterjee, M. J. Org. Chem. 1995, 60, 5958–5960. doi:10.1021/jo00123a037 |
| 17. | Mu, G.; Pu, W.; Zhou, M.; Liu, Y.; Yang, H.; Wang, C. Chin. J. Org. Chem. 2013, 33, 1298–1303. doi:10.6023/cjoc201303016 |
| 21. | Daskiewicz, J.-B.; Depeint, F.; Viornery, L.; Bayet, C.; Comte-Sarrazin, G.; Comte, G.; Gee, J. M.; Johnson, I. T.; Ndjoko, K.; Hostettmann, K.; Barron, D. J. Med. Chem. 2005, 48, 2790–2804. doi:10.1021/jm040770b |
| 22. | Al-Maharik, N.; Botting, N. P. Tetrahedron 2003, 59, 4177–4181. doi:10.1016/S0040-4020(03)00579-9 |
| 23. | Poerwono, H.; Sasaki, S.; Hattori, Y.; Higashiyama, K. Bioorg. Med. Chem. Lett. 2010, 20, 2086–2089. doi:10.1016/j.bmcl.2010.02.068 |
© 2015 Mei et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)