Chemistry Department “G. Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 1, 20133 Milano, Italy
Corresponding author email
Associate Editor: S. Bräse Beilstein J. Org. Chem.2015,11, 2117–2124.https://doi.org/10.3762/bjoc.11.228 Received 13 Jul 2015,
Accepted 22 Oct 2015,
Published 05 Nov 2015
The first stereoselective synthesis of lippidulcines A, B and C has been accomplished starting from (+)-hernandulcin, which has been prepared on a multigram scale. The previously assigned absolute configurations have been confirmed. The key steps of this synthesis are based on a modified version of the Kornblum–DeLaMare rearrangement, and on a highly regioselective and stereoselective ketone reduction with the MeCBS reagent. The taste evaluations indicate that none of these sesquiterpenes are sweet, instead the lippidulcine A is a cooling agent with a mint after taste.
It is a matter of fact that a large consumption of sucrose is strongly associated to a considerable number of undesirable health effects, among which cardiovascular diseases and dental caries are the most relevant. Moreover, the increasing number, especially in the western countries, of people with obesity and type-II diabetes has pushed the food industry to develop new low calorie sweeteners, better known as sugar substituents. Among all artificial sweeteners so far developed: aspartame, saccharin, acesulfame K and sucralose are undoubtedly the most popular. However, questions regarding the safety of these sweeteners are still largely argued from the scientific community [1]. Thus, the discovery of new sugar substituents has become a target of food industry, to this regard new sweet-tasting natural products might offer a valid alternative to the artificial ones [2-4].
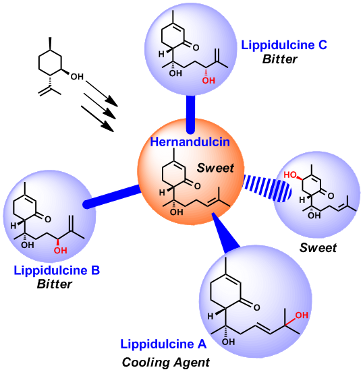
At the beginning of 1980s Kinghorn et al. came across with an ancient botanical report describing the existence of a New World plant with comestible leaves having a very intense sweet taste. The Aztecs used to call this plant “Tzonpelic xihuitl”, which means sweet herb. However, for several centuries its unusual property has been incredibly forgotten until 1985; when the sesquiterpene 1 (Figure 1), isolated from the leaves and flowers of the Mexican plant Lippia dulcis, resulted the main vector of sweetness that Hernández described four hundred years before in his treatise [5]. Thus, in honor of the Spanish scientist, this natural product was called hernandulcin [6]. Hernandulcin is the first strongly sweet sesquiterpene, making it a breakthrough discovery in the field of natural sugar substitutes. It turned out that 1, which belongs to the family of bisabolanes, is more than 1000 times as sweet as sucrose.
Figure 1:
Hernandulcin and other bisabolanic derivatives extracted from Lippia dulcis.
Figure 1:
Hernandulcin and other bisabolanic derivatives extracted from Lippia dulcis.
More recently other derivatives of 1 have been discovered, i.e., the peroxylippidulcines 2a–c and the lippidulcines 3a–c (Figure 1) [7,8]. However, these sesquiterpenes have been isolated in a so small amount that it has not been possible to assess their taste. In principle these bisabolanes could represent a very interesting structural variation of 1, especially the lippidulcines, since the presence of a second hydroxy group should increase significantly their solubility in water, which is a key property for the sweeteners of beverages. In the following we report on the synthesis of hernadulcin and its hydroxy derivatives, i.e., the lippidulcines A, B and C and their taste evaluation.
Results and Discussion
Since our synthetic strategy for the preparation of lippidulcines 3a–c is based on the photooxygenation of 1, it was mandatory to develop and optimize the synthesis of the latter on a multigram scale. Few years ago we have reported a new stereospecific synthesis of (+)-hernandulcin [9] starting from the very cheap commercially available (+)-isopulegol.
Mori [10,11] and Cheon [12,13] reported some syntheses of 1. They epoxidized the side-chain olefinic double bonds of (+)-limonene or (−)-isopulegol with m-chloroperbenzoic acid (MCPBA) to introduce the stereogenic center at the C(2’) position. Unfortunately both methods were only modestly stereoselective and resulted in mixtures of the respective epoxides. These results implied a not simple column chromatographic separation of the diastereomeric mixtures. In contrast, we have shown that the Katsuki–Sharpless epoxidation of (+)-neoisopulegol (4) is much more selective [9].
In the following we describe an improved version of our previously reported synthesis of 1. First we have focused our initial efforts on the synthesis optimisation of the key intermediate 4 that can be prepared starting from (−)-isopulegol by two different approaches: i) oxidation of the hydroxy group to give (S)-isopulegone, which in turn is reduced stereospecifically into the desired cis diastereoisomer; or ii) by inversion of C(1) stereogenic center by means of a Mitsunobu reaction, followed by transesterification of the ester to give 4 (Scheme 1).
Scheme 1:
Synthesis of (+)-neoisopulegol. Reagents and conditions: (a) Jones reagent, acetone, 0 °C, 3 h; (b) DIBALH, 2-propanol, toluene, 0 °C/rt, 6 h; (c) L-selectride, THF, −78 °C/rt, 16 h; (d) ADH, cofactor NAD(P)H+, GDH, glucose, water, 30 °C, 24 h; (e) p-nitrobenzoic acid, PPh3, DIAD, toluene, 0 °C, 24 h; (f) MeONa, MeOH, rt, 5 h.
Scheme 1:
Synthesis of (+)-neoisopulegol. Reagents and conditions: (a) Jones reagent, acetone, 0 °C, 3 h; (b)...
(S)-Isopulegone was prepared by Jones oxidation of (−)-isopulegol following a reported procedure [14]; but on a large scale we have observed that a partial loss of the optical purity of the product might easily occur during the reaction. Then, we tested three different reducing agents (methods A, B and C). Indeed, even if the reduction with an over stoichiometric amount of L-selectride at −78 °C in THF gave excellent results (method A) [9], since 4 was isolated in an 84% yield and with a de of 97% [9], this reagent is quite expensive hampering its utilization on a large scale. For this reason we tried the more convenient Meerwein–Ponndorf–Verley reduction [15] (DIBAL-H, iPrOH in toluene at room temperature), but both the yield and the selectivity did not result satisfactory (10% conversion, 0% de, method B). Undoubtedly, a catalytic and a more operationally simple procedure would be highly desirable, to this regard the enzymatic reduction of isopulegone was tested as well (method C). Thus, the biocatalysed reduction [16] of this ketone with a panel of commercially available alcohol dehydrogenases (ADHs) was screened [17], the regeneration of the NAD(P)+ cofactor was carried out using a glucose dehydrogenase (GDH from Bacillus megaterium), with glucose as co-substrate [18]. The product distributions are reported in Table 1. However, most of these ADHs gave low conversions and selectivity. The best performances were obtained with the Thermoanaerobium brokii ADH; but even if the de was excellent (>99%) the conversion was still too low (about 36% by GC) to be really exploited on a preparative scale.
Table 1:
Alcohol dehydrogenase screening results, method C.
ADH source
(S)-isopulegone [%]a
(−)-isopulegol [%]a
4 [%]a
cofactor
de [%]a
Candida parapsilosis
100.0
0
0
NAD+
–
Rhodococcus erythropolis
98.5
0.5
1.0
NAD+
29.3
Baker's yeast
100
0
0
NAD+
–
Horse liver recombinant
100
0
0
NAD+
–
Thermoanaerobium brockii
63.7
0.1
36.2
NADP+
99.7
Lactobacillus kefir
90.5
9.5
0
NADP+
−99.9
Parvibaculum lavamentivorans
94.4
0.5
5.2
NADP+
82.5
Deinococcus radiodurans
100
0
0
NADP+
–
Ketoreductase
73.7
15.5
10.8
NADP+
−17.9
aBy GC–MS.
Finally, we tried the Mitsunobu reaction, which gave the best results (method D) [19]. Indeed, the treatment of (–)-isopulegol with diisopropyl azodicarboxylate (DIAD) and p-nitrobenzoic acid in the presence of triphenylphosphine gave the corresponding ester in an almost quantitative yield. The latter was easily purified by crystallization from n-hexane, and then transesterificated with MeONa giving 4 in an overall yield of 84% and with an excellent de of >99%, by GC–MS ([α]D +25° (c 2.0, CHCl3) vs lit. distomer (−)-neoisopulegol [19] [α]D −22.2° (c 2.0, CHCl3) or (+)-neoisopulegol [20] [α]D +28.7° (c 17.2, CHCl3)).
The subsequent synthesis of 1 shown Scheme 2 is a slightly adapted version of the synthesis published in reference [9].
First, the metallation [21] of 4 with t-BuOK and n-BuLi in hexane followed by addition of prenyl bromide (3-methyl-2-butenyl bromide) at −10 °C afforded the sesquiterpene derivative 5 ([α]D +19.5° (c 1.2, CHCl3)) in 84% yield. The VO(acac)2-catalyzed epoxidation of 5 with tert-butylhydroperoxide (TBHP) in toluene gave 6 ([α]D +34.2° (c 1.3, CHCl3)) in 95% yield [9,22]. Then, the diol 7 was obtained in 85% yield from the reduction of epoxide 6 with LiAlH4 in THF at 0 °C. The diol 7 was easily purified by crystallization (85% yield, n-hexane at −50 °C up to −30 °C). Treatment of 7 with Dess–Martin periodinane (DMP) [23] in CH2Cl2 at room temperature gave ketone 8 ([α]D +11.0° (c 1.0, CHCl3)) in 90% yield.
The most difficult step of this synthetic route is the dehydrogenation of ketone 8. Since our main interest is focused on the taste evaluation of the final products, we deliberately avoided all toxic selenium based reagents [12,13]. We tested several methodologies and the results are summarized in Table 2.
Table 2:
Investigated methodologies for preparation of 1.
Substance
Conversion [%]a
Protocol
8
–
IBX, DMSO, 80 °C, 12 hb
10
trace
IBX, MPO, DMSO, rt, 12 hb
10
54
Pd(OAc)2, O2, DMSO, 58 °C, 3 db
10
21
Pd(TFA)2, O2, Na2HPO4, MeCN, rt, 12 h
10
6
Pd(OAc)2, Oxone, Na2HPO4, MeCN, 50 °C, 38 h
10a
trace
Pd(TFA)2, O2, Na2HPO4, DMSO/CH2Cl2 2:1, rt, 1 d
10a
4
Pd(OAc)2, O2, MeCN/CH2Cl2 2:1, 50 °C, 7 d
10a
–
Pd(OAc)2, dppe, diallyl carbonate, MeCN/CH2Cl2 2:1, 50 °C, 12 h
aBy GC–MS; bThese methods have been previously tested [9].
In our initial synthesis [9] we applied the hyperiodine chemistry, developed by Nicolaou. However, the o-iodoxybenzoic acid (IBX) mediated oxidation of 8 to give the enone 1 in DMSO at high temperature (80 °C) resulted unsuccessful [24]. The hydrogenation of silyl enol ether derivatives in the presence of the IBX-N-oxide complex gives the corresponding enones, usually with better conversion and under milder conditions (room temperature) [25]. Thus, first the tertiary alcohol of 8 was protected as trimethylsilyl ether giving 9 ([α]D −11.5° (c 1.3, CHCl3), vs lit. [13] [α]D −16.3° (c 0.12, EtOH)), which, after treatment with lithium diisopropylamide (LDA) followed by addition of trimethylsilyl chloride (TMSCl) at –78 °C in THF afforded the kinetic enol ether 10 in 91% yield ([α]D +19.7° (c 1.4, CHCl3)). The latter was submitted to the oxidative step with the IBX-N-oxide, but even in this case the results were unsatisfactory since only traces of enone 11 were detected. In contrast, the Pd based Saegusa–Larock methodology resulted successful [26,27], indeed when 10 was treated with a substoichiometric amount of Pd(OAc)2 in DMSO at 58 °C under an oxygen atmosphere, it was possible to isolate 11 ([α]D +11.1° (c 1.4, CHCl3), vs lit. [13] [α]D +9.7° (c 0.14, EtOH)) in a maximum yield of 54%, as no starting material was present anymore in the reaction mixture.
Very recently, Stahl et al. have proved that by replacing Pd(OAc)2 with Pd(TFA)2 it is possible to dehydrogenate directly the ketones at room temperature [28], but without the possibility of controlling the regioselectivity. Since, in our case this issue is critical we tried to apply the Pd(TFA)2 catalyzed dehydrogenation on the silyl enol ether 10 and in the presence of Na2HPO4 buffer, in order to mitigate the detrimental acidity of TFA. However, 11 was produced in a modest yield of 21%, because, even at these mild conditions, 10 reconverted to the initial ketone 9 faster than its oxidative dehydrogenation. Then we tested another procedure, in which the co-oxidant O2 was replaced with Oxone [29], but even in this case the conversion (6% by GC) was worse than that achieved using bubbling O2. Further attempts of optimizing the oxidative dehydrogenative step of the Stahl protocol were carried out by changing the trimethylsilyl enol ether group with the more robust triethylsilyl enol ether, 10a. In principle this enolether should be more compatible with the Pd(TFA)2 catalyst, but unfortunately 10a is insoluble in the typical solvents in which are carried out these Pd-catalyzed dehydrogenations (mainly DMSO or MeCN); even when using mixed co-solvent systems, the results were still very poor. Next, we tested the Tsuji variant [30] (Pd(OAc)2, dppe, diallyl carbonate, MeCN) but both 10 and 10a decomposed during the reaction.
Finally, (+)-hernandulcin 1 was obtained in a 92% yield by cleavage of the silyl protective group of 11 with tetra-n-butylammonium fluoride (TBAF) in MeCN and at room temperature, the spectroscopic data of 1 were in complete agreement with those reported in literature [6] ([α]D +130° (c 1.6, CHCl3) vs lit. [31] ([α]D +115° (c 0.64, CHCl3)).
The photooxygenation [32] of (+)-hernandulcin in a mixed solvent system (CH2Cl2/MeOH, 4:1) and in presence of a catalytic amount of the methylene blue photosensitizer (<0.1% in weight, λ = 280 nm) afforded a mixture of peroxylippidulcines A, B and C, i.e., 2a–c, in the ratio of 47:21:32 (by 1H NMR) and in an almost quantitative yield (Scheme 3). However, any attempt of isolating each isomer of peroxylippidulcine by column chromatography technique failed. In addition, the singlet dark oxidation (H2O2 with a catalytic amount of Na2MoO4 at 55 °C) was tested, but without success [33].
Then, we tried to separate 2a–c by means of a regioselective O-silylation of the secondary hydroperoxide with the tert-butyldiphenylsilyl chloride (TBDPSiCl), but with our surprise most of the starting material was consumed, and after column chromatography separation only the O-silylated derivative of peroxylippidulcine A, 12, and a small amount of ketone 13 were isolated (Scheme 3). The latter is likely produced by a variant of the Kornblum–DeLaMare rearrangement [34,35] of the O-silyl precursor at room temperature, unfortunately 13 is very unstable. However, we envisaged the possibility of using this ketone, prior its tertiary alcohol protection, as a key-precursor for the preparation of lippidulcines B and C by means of a stereoselective and regioselective reduction of the exocyclic carbonyl group C(1). Indeed, the peroxy lippidulcines are not interesting from the sensorial point of view, since the taste of 2a ([α]D +43.5° (c 1.6, CHCl3) vs lit. [8] [α]D +42.0° (c 3.2, CHCl3)), obtained by treatment of 12 with TBAF in MeCN, resulted very bitter, as usually are the hydroperoxides (Table 3).
Thus, the crude material of the photooxygenation was treated with an over stoichiometric amount of TMSCl (Scheme 3). In these conditions, after 24 hours most of peroxylippidulcine A was completely O-silylated, whereas the B and C ones rearranged partially to give the O-silyl protected ketone, 14 ([α]D +13.6° (c 1.1, CHCl3). Since in this case, the Kornblum–DeLaMare rearrangement was not quantitative (around 68% by 1H NMR), after the aqueous work-up followed by treatment of the reaction mixture with triphenylphosphine, the unreacted lippidulcines B and C were oxidized with MnO2 to give ketone 14 together to the unreacted O-TMS protected lippidulcine A, 15a ([α]D −11.3° (c 1.0, CHCl3)). The latter were easily separated by column chromatography. Then, the O-silyl group of 15a was cleaved with TBAF affording lippidulcine A ([α]D +132° (c 1.3, CHCl3) vs lit. [8] [α]D +123.6° (c 0.1, CHCl3)), which turned out to be a very pleasant cooling agent with a very light mint after taste (Table 3).
Ketone 14 was submitted to the Corey–Bakshi–Shibata stereoselective carbonyl reduction protocol [36] using (S)-MeCBS as catalyst (1.2 equiv) in CH2Cl2, and in the presence of an over stoichiometric amount of the Me2S.BH3 complex (Scheme 4). After column chromatography, the reduced product 15b ([α]D −1.6° (c 0.8, CHCl3)) was obtained in a good yield of 80%.
Scheme 4:
Reagents and conditions: (a) (S)-MeCBS or (R)-MeCBS for 15b or 15c, respectively, BH3·Me2S, −78 °C for 1 d then −60 °C for 2 d; (b) TBAF, MeCN, rt, 30 min.
Scheme 4:
Reagents and conditions: (a) (S)-MeCBS or (R)-MeCBS for 15b or 15c, respectively, BH3·Me2S, −78 °C ...
Thus, lippidulcine B (3b), after TBAF mediated cleavage of the silyl protecting group, was isolated in 73% yield and with an excellent de of 99% (by 1H NMR), ([α]D +123° (c 1.5, CHCl3) vs lit. [8] [α]D +113.3° (c 0.4, CHCl3)), confirming the absolute stereochemistry that it has been previously assigned. It is noteworthy that in this case the Corey asymmetric reduction of the carbonyl group is completely regioselective in favour of the exocyclic carbonyl group. To knowledge of the authors, just another example has been reported with a similar regioselectivity [37]. Then, lippidulcine C ([α]D +92.1° (c 1.1, CHCl3) vs lit. [8] [α]D +119.8° (c 0.7, CHCl3)) was prepared following the same route adopted for the synthesis of 3b, but using the (R)-MeCBS.
Finally, the samples of lippidulcines were submitted to the taste evaluation, the results are summarized in Table 3.
Table 3:
Sensorial evaluation of lippidulcines.
Substance
taste descriptiona
2a
Very bitter with a light mint after taste
3a
Pleasant cooling agent, fresh sensation with a very slight mint after taste
3b
Fresh pungent then bitter
3c
Bitter
3d
Slightly fresh pungent, then very bitter
aThe evaluation has been made on a panel of 4 people.
In summary, the introduction of an hydroxy group on the side chain of hernandulcin has changed drastically the taste of the latter, indeed the lippidulcines B and C are bitter, whereas, very surprisingly 3a turned out to be a new natural cooling agent [38] with a light mint retro taste. These results are in contrast with the behavior of the (+)-β-hydroxyhernadulcin isomer (Figure 1), which has been described as a sweetener [39]. Intrigued by the behavior of 3a we prepared the epimer 3d ([α]D −107° (c 1.2, CHCl3) vs lit. [8] [α]D −118.4° (c 0.5, CHCl3)) by the acid catalyzed racemization of C(6) stereogenic center. Remarkably, the absolute configuration at this stereocenter plays in important role, since 3d resulted pungent cool and bitter.
Conclusion
We improved the synthesis of (+)-hernadulcin on a multigram scale, and we have accomplished the first total synthesis of peroxylippidulcine A and lippidulcines A, B and C, confirming their absolute stereochemical configurations that have been previously assigned. The key steps are: i) a modified version of the Kornblum–DeLaMare rearrangement, promoted by the O-silylation of the hydroperoxy group, and ii) an highly regioselective and stereoselective reduction of the exocyclic carbonyl group of ketone 14 with the Corey reagent. The taste evaluation of these bisabolanes has demonstrated that the insertion of a hydroxy group on the side chain of hernadulcin annuls its intense sweetness.
Supporting Information
Supporting Information File 1:
Experimental procedures, analytical data, and copies of 1H and 13C NMR spectra of all compounds.
We are grateful to Dr. Stefano Serra of ICRM (Milan) for the very fruitful discussion of results. We thank Michele Crotti for the preliminary work with the ADHs.
References
Weihrauch, M. R.; Diehl, V. Ann. Oncol.2004,15, 1460–1465. doi:10.1093/annonc/mdh256
Return to citation in text:
[1]
Kinghorn, A. D.; Soejarto, D. D. Pure Appl. Chem.2002,74, 1169–1179. doi:10.1351/pac200274071169
Return to citation in text:
[1]
Kinghorn, A. D.; Chin, Y.-W.; Pan, L.; Jia, Z. In Comprehensive Natural Products Chemistry II: Chemistry and Biology; Verpoorte, R., Ed.; Development & Modification of Bioactivity, Vol. 3; Elsevier: Oxford, U.K., 2010; pp 269–315.
Return to citation in text:
[1]
Behrens, M.; Meyerhof, W.; Hellfritsch, C.; Hofmann, T. Angew. Chem., Int. Ed.2011,50, 2220–2242. doi:10.1002/anie.201002094
Return to citation in text:
[1]
This treatise is entitled “Quatro libros de la naturaleza y virtudes de las plantas y animales que estan receuidos en el uso de la medicina en la Nueva España”.
Return to citation in text:
[1]
Compadre, C. M.; Pezzuto, J. M.; Kinghorn, A. D.; Kamath, S. K. Science1985,227, 417–419. doi:10.1126/science.3880922
Return to citation in text:
[1]
[2]
Ono, M.; Morinaga, H.; Masuoka, C.; Ikeda, T.; Okawa, M.; Kinjo, J.; Nohara, T. Chem. Pharm. Bull.2005,53, 1175–1177. doi:10.1248/cpb.53.1175
Return to citation in text:
[1]
Ono, M.; Tsuru, T.; Abe, H.; Eto, M.; Okawa, M.; Abe, F.; Kinjo, J.; Ikeda, T.; Nohara, T. J. Nat. Prod.2006,69, 1417–1420. doi:10.1021/np068010m
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
Bahia, P. S.; Jones, M. A.; Snaith, J. S. J. Org. Chem.2004,69, 9289–9291. doi:10.1021/jo049300f
Return to citation in text:
[1]
Nakamura, K.; Yamanka, R.; Matsuda, T.; Harada, T. Tetrahedron: Asymmetry2003,14, 2659–2681. doi:10.1016/S0957-4166(03)00526-3
Return to citation in text:
[1]
Brenna, E.; Gatti, F. G.; Monti, D.; Parmeggiani, F.; Sacchetti, A. ChemCatChem2012,4, 653–659. doi:10.1002/cctc.201100418
Return to citation in text:
[1]
Bechtold, M.; Brenna, E.; Femmer, C.; Gatti, F. G.; Panke, S.; Parmeggiani, F.; Sacchetti, A. Org. Process Res. Dev.2011,16, 269–276. doi:10.1021/op200085k
Return to citation in text:
[1]
Yadav, J. S.; Bhasker, E. V.; Srihari, P. Tetrahedron2010,66, 1997–2004. doi:10.1016/j.tet.2010.01.054
Return to citation in text:
[1]
[2]
Moreira, J. A.; Corrêa, A. G. Tetrahedron: Asymmetry2003,14, 3787–3795. doi:10.1016/j.tetasy.2003.09.030
Return to citation in text:
[1]
Ito, Y.; Hirao, T.; Saegusa, T. J. Org. Chem.1978,43, 1011–1013. doi:10.1021/jo00399a052
Return to citation in text:
[1]
Larock, R. C.; Hightower, T. R.; Kraus, G. A.; Hahn, P.; Zheng, D. Tetrahedron1995,36, 2423–2426. doi:10.1016/0040-4039(95)00306-W
Return to citation in text:
[1]
Diao, T.; Stahl, S. S. J. Am. Chem. Soc.2011,133, 14566–14569. doi:10.1021/ja206575j
Return to citation in text:
[1]
Lu, Y.; Nguyen, P. L.; Lévaray, N.; Lebel, H. J. Org. Chem.2014,78, 776–779. doi:10.1021/jo302465v
Return to citation in text:
[1]
Tsuji, J.; Minami, L.; Shimizu, I. Tetrahedron Lett.1983,24, 5635–5638. doi:10.1016/S0040-4039(00)94160-1
Return to citation in text:
[1]
Souto-Bachiller, F. A.; De Jesus-Echevarria, M.; Cárdenas-González, O. E.; Acuña-Rodriguez, M. F.; Meléndez, P. A.; Romero-Ramsey, L. Phytochemistry1997,44, 1077–1086. doi:10.1016/S0031-9422(96)00691-7
Return to citation in text:
[1]
Alsters, L. P.; Jary, W.; Nardello-Rataj, V.; Aubry, J.-M. Org. Process Res. Dev.2010,14, 259–262. doi:10.1021/op900076g
Return to citation in text:
[1]
Kornblum, N.; DeLaMare, H. E. J. Am. Chem. Soc.1951,73, 880–881. doi:10.1021/ja01146a542
Return to citation in text:
[1]
Zheng, X.; Lu, S.; Li, Z. Org. Lett.2013,15, 5432–5435. doi:10.1021/ol402509u
Return to citation in text:
[1]
Corey, E. J.; Shibata, S.; Bakshi, R. K. J. Org. Chem.1988,53, 2861–2863. doi:10.1021/jo00247a044
Return to citation in text:
[1]
Moustafa, G. A. I.; Kamada, Y.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem.2012,10, 8609–8615. doi:10.1039/c2ob26532j
Return to citation in text:
[1]
Serra, S.; Fuganti, C.; Gatti, F. G. Eur. J. Org. Chem.2008, 1031–1037. doi:10.1002/ejoc.200701010
Return to citation in text:
[1]
Kaneda, N.; Lee, I.-S.; Gupta, M. P.; Soejarto, D. D.; Kinghorn, A. D. J. Nat. Prod.1992,55, 1136–1141. doi:10.1021/np50086a016
Return to citation in text:
[1]
This treatise is entitled “Quatro libros de la naturaleza y virtudes de las plantas y animales que estan receuidos en el uso de la medicina en la Nueva España”.
Kinghorn, A. D.; Chin, Y.-W.; Pan, L.; Jia, Z. In Comprehensive Natural Products Chemistry II: Chemistry and Biology; Verpoorte, R., Ed.; Development & Modification of Bioactivity, Vol. 3; Elsevier: Oxford, U.K., 2010; pp 269–315.
4.
Behrens, M.; Meyerhof, W.; Hellfritsch, C.; Hofmann, T. Angew. Chem., Int. Ed.2011,50, 2220–2242. doi:10.1002/anie.201002094
Souto-Bachiller, F. A.; De Jesus-Echevarria, M.; Cárdenas-González, O. E.; Acuña-Rodriguez, M. F.; Meléndez, P. A.; Romero-Ramsey, L. Phytochemistry1997,44, 1077–1086. doi:10.1016/S0031-9422(96)00691-7

![[1860-5397-11-228-1]](/bjoc/content/figures/1860-5397-11-228-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-228-i1]](/bjoc/content/inline/1860-5397-11-228-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-228-i5.svg?max-width=637&scale=1.0)
![[1860-5397-11-228-i2]](/bjoc/content/inline/1860-5397-11-228-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-228-i3]](/bjoc/content/inline/1860-5397-11-228-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-228-i4]](/bjoc/content/inline/1860-5397-11-228-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
