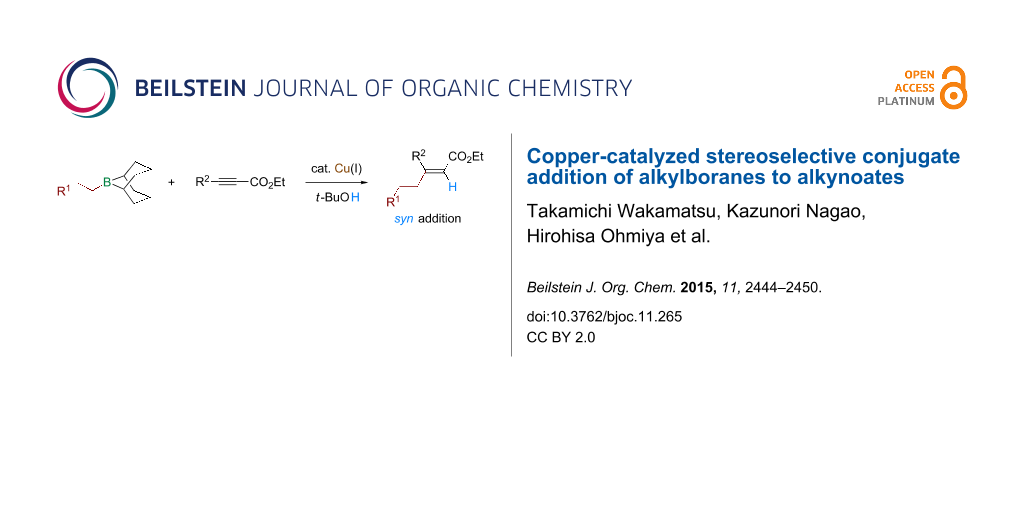
Abstract
A copper-catalyzed conjugate addition of alkylboron compounds (alkyl-9-BBN, prepared by hydroboration of alkenes with 9-BBN-H) to alkynoates to form β-disubstituted acrylates is reported. The addition occurred in a formal syn-hydroalkylation mode. The syn stereoselectivity was excellent regardless of the substrate structure. A variety of functional groups were compatible with the conjugate addition.

Graphical Abstract
Introduction
Copper-mediated conjugate additions of organometallic reagents to alkynoates are powerful tools for the synthesis of multisubstituted alkenes [1-8]. Because of their broad availability and their compatibility with a multitude of functional groups, organoboron compounds are especially popular organometallic reagents. Recently, Yamamoto and co-workers developed copper-catalyzed conjugate additions of aryl- and allylboron compounds to alkynoates [9,10], but alkylboron compounds have not been used for these methods [11].
As related studies we reported earlier the copper-catalyzed conjugate addition of alkylboranes (alkyl-9-BBN) to imidazole-2-yl α,β-unsaturated ketones [12-14] and the copper-catalyzed three-component coupling with alkylboranes, alkynoates, and tributyltin methoxide to form trisubstituted alkenylstannanes [15]. The latter reaction pathway involved Sn-trapping of an alkenylcopper intermediate that was formed through syn-carbocupration of an alkylcopper(I) species across the C–C triple bond of the alkynoate. We envisioned that 1,2-hydroalkylation of the alkynoates might be possible through proton-trapping of an alkenylcopper intermediate.
Herein, we report a copper-catalyzed conjugate addition of alkylboranes to alkynoates, providing a versatile approach to β-disubstituted acrylates [16-19]. The addition occurred in a formal syn-hydroalkylation mode. The syn stereoselectivity was excellent regardless of the substrate structure, and a variety of functional groups were tolerated in both the alkylborane and the alkynoate.
Results and Discussion
Alkylborane 2a (0.275 mmol), which was obtained via hydroboration of styrene (1a) with the 9-borabicyclo[3.3.1]nonane (9-BBN-H) dimer, and ethyl 3-phenylpropiolate (3a, 0.25 mmol) were treated with CuOAc (5 mol %), t-BuOK (5 mol %), P(OPh)3 (10 mol %), and t-BuOH (0.25 mmol) in 1,4-dioxane (1.2 mL) at 40 °C for 12 h. The reaction afforded a formal hydroalkylation product, β-disubstituted acrylate 4aa in 99% isolated yield with >99% syn selectivity (Scheme 1).
![[1860-5397-11-265-i1]](/bjoc/content/inline/1860-5397-11-265-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Conjugate addition of alkylborane 2a to alkynoate 3a.
Scheme 1: Conjugate addition of alkylborane 2a to alkynoate 3a.
The results of ligand screening for the reaction between 2a and 3a are summarized in Table 1. P(OPh)3 was the most effective ligand in terms of product yield and syn selectivity (Table 1, entry 1). The use of other monophosphine ligands such as PPh3 and PCy3 or the DPPE bisphopshine was also effective in promotion of the reaction, but resulted in a reduced stereoselectivity (Table 1, entries 2–4). No reaction occurred with N-heterocyclic carbenes (NHC) such as IMes or IPr (Table 1, entries 5 and 6). The reaction with (IMes)CuCl or (IPr)CuCl complex delivered no reaction product (data not shown). The reaction without a ligand resulted in a significantly decreased product yield while the syn selectivity was fairly high (Table 1, entry 7).
Table 1: Ligand effects.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-265-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Liganda | Yield [%]b | syn/antic |
|---|---|---|---|
| 1 | P(OPh)3 | 99 | >99:1 |
| 2 | PPh3 | 99 | 67:33 |
| 3 | PCy3 | 56 | 64:36 |
| 4 | DPPE | 99 | 83:17 |
| 5 | IMes | 0 | – |
| 6 | IPr | 0 | – |
| 7 | none | 37 | 97:3 |
aIMes: 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene, IPr: 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene. bYield determined by 1H NMR. cDetermined by 1H NMR or GC analysis of the crude product.
The use of less expensive CuCl as a copper salt was also effective to produce 4aa in 90% yield with 99% syn selectivity. The reaction using MeOH as a proton source instead of t-BuOH caused a drastic reduction in the product yield with the syn selectivity slightly decreased (38%, syn/anti 97:3). The reduction of the yield might be due to the protonation of an alkylcopper species by the more acidic MeOH (vide infra). There was no reaction in the absence of a proton sourse. No hydroalkylation product at all could be found when alkyl-9-BBN 2a was replaced by (2-phenylethyl)boronic acid pinacolate ester; the substrates hardly reacted at all.
A variety of β-disubstituted acrylates were accessible through the hydroboration–conjugate addition one-pot protocol with excellent syn stereoselectivities (Table 2). This protocol tolerated functional groups such as methoxy, ester, phthalimide, fluoro, cyano and aldehyde moieties in the alkylboranes and alkynoates (Table 2, entries 1–3, 6–9 and 11).
Table 2: Copper-catalyzed conjugate addition of alkylboranes 2 to alkynoates 3.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-265-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Alkene | Alkynoate | Product | Yield [%]b | syn/antic |
|---|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-265-i6.svg?max-width=637&scale=1.0)
1b |
3a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-265-i7.svg?max-width=637&scale=1.0)
4ba |
94 | 99:1 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-265-i8.svg?max-width=637&scale=1.0)
1c |
3a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-265-i9.svg?max-width=637&scale=1.0)
4ca |
99 | >99:1 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-265-i10.svg?max-width=637&scale=1.0)
1d |
3a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-265-i11.svg?max-width=637&scale=1.0)
4da |
87 | 99:1 |
| 4d |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-265-i12.svg?max-width=637&scale=1.0)
1e |
3a |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-265-i13.svg?max-width=637&scale=1.0)
4ea |
86 | >99:1 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-265-i14.svg?max-width=637&scale=1.0)
1f |
3a |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-265-i15.svg?max-width=637&scale=1.0)
4fa |
0 | – |
| 6 | 1a |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-265-i16.svg?max-width=637&scale=1.0)
3b |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-265-i17.svg?max-width=637&scale=1.0)
4ab |
96 | 98:2 |
| 7 | 1a |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-265-i18.svg?max-width=637&scale=1.0)
3c |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-265-i19.svg?max-width=637&scale=1.0)
4ac |
91 | 99:1 |
| 8 | 1a |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-265-i20.svg?max-width=637&scale=1.0)
3d |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-265-i21.svg?max-width=637&scale=1.0)
4ad |
88 | 99:1 |
| 9 | 1a |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-265-i22.svg?max-width=637&scale=1.0)
3e |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-265-i23.svg?max-width=637&scale=1.0)
4ae |
93 | 99:1 |
| 10 | 1a |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-265-i24.svg?max-width=637&scale=1.0)
3f |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-265-i25.svg?max-width=637&scale=1.0)
4af |
95 | 94:6 |
| 11 | 1a |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-265-i26.svg?max-width=637&scale=1.0)
3g |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-265-i27.svg?max-width=637&scale=1.0)
4ag |
90 | >99:1 |
| 12 | 1a |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-265-i28.svg?max-width=637&scale=1.0)
3h |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-265-i29.svg?max-width=637&scale=1.0)
4ah |
94 | >99:1 |
| 13 | 1a |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-265-i30.svg?max-width=637&scale=1.0)
3i |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-265-i31.svg?max-width=637&scale=1.0)
4ai |
98 | >99:1 |
| 14 | 1a |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-265-i32.svg?max-width=637&scale=1.0)
3j |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-265-i33.svg?max-width=637&scale=1.0)
4aj |
93 | >99:1 |
aThe reaction was carried out with 3 (0.25 mmol), 2 (0.275 mmol), CuOAc (5 mol %), t-BuOK (5 mol %), P(OPh)3 (10 mol %) and t-BuOH (0.25 mmol) in dioxane (1.2 mL) at 40 °C for 12 h. Alkylborane 2 was prepared in advance by hydroboration of 1 with the 9-BBN-H dimer at 60 °C for 1 h and used without purification. bYield of isolated product. cDetermined by 1H NMR or GC analysis of the crude product. dDiasteremeric ratio (1:1).
The data in Table 2 show the variety of functional groups attached to alkylboranes 2 that are tolerated in the reaction. The rather crowded alkylborane 2c, which was prepared from tertiary alkyl substituted terminal alkene 1c, reacted nicely (Table 2, entry 2). β-Branched alkylborane 2e, prepared from α-methylstyrene (1e), provided 4ea in good yield (Table 2, entry 4). Unfortunately, however, the reaction of secondary alkylboranes made from internal alkenes, did not work (Table 2, entry 5).
The variety of alkynoates used is also shown in Table 2. The fluoro atom and the methoxy, cyano and aldehyde groups were acceptable as para or meta-substituents on the aromatic ring at the β-positions (Table 2, entries 6–9). The alkynoate 3f bearing a 2-thienyl group at the β-position is also compatible with the conjugate addition and gave 94% syn selectivity (Table 2, entry 10). The 1,3-enyne derivative 3g reacted regioselectively to afford a conjugated 2,4-dienoate 4ag in 90% yield with excellent syn selectivity (Table 2, entry 11).
Alkyl groups were also acceptable as β-substituent of the alkynoates (Table 2, entries 12–14). Alkynoate 3h with a methyl group at the β-position reacted with an excellent stereoselectivity (Table 2, entry 12). The alkynoates with an ethyl (3i) or cyclohexyl group (3j) were also suitable substrates (Table 2, entries 13 and 14).
Alkene hydroboration of 1k followed by copper-catalyzed intramolecular conjugate addition enabled the formation of the corresponding five-membered carbocycle 4k in 94% yield with complete syn selectivity (Scheme 2).
![[1860-5397-11-265-i2]](/bjoc/content/inline/1860-5397-11-265-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of five membered carbocycle.
Scheme 2: Synthesis of five membered carbocycle.
To gain insight into the mechanism of the copper-catalyzed conjugate addition, the reaction between 2a and 3a with t-BuOD under the optimum conditions was conducted (Scheme 3). The reaction afforded 4aa-D, which is deuterated at the α-position of the carbonyl group (93% D). The syn selectivity was slightly decreased due to the deuterium isotope effect: Slower D-trap caused isomerization of organocopper intermediates (vide infra). This experimental result indicates that t-BuOH acts as a proton source.
![[1860-5397-11-265-i3]](/bjoc/content/inline/1860-5397-11-265-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
A possible mechanism for the present copper catalysis is proposed in Figure 1. An alkoxycopper complex (A) is initially formed by the reaction of CuOAc, t-BuOK and P(OPh)3. Boron-to-cupper transmetalation between A and the alkylborane 2 occurs to form an alkylcopper(I) species (B) and a t-butoxyborane (9-BBN-Ot-Bu) [12-15,20-25]. Subsequently, the alkylcopper species B forms a π-complex (C) with alkynoate 3. Then, syn-carbocupration across the C–C triple bond of C with the assistance of Lewis acidic activation with the tert-butoxyborane gave an alkenylcopper intermediate (D). Finally, protonolysis by t-BuOH produces syn-4, regenerating the alkoxycopper complex A.
![[1860-5397-11-265-1]](/bjoc/content/figures/1860-5397-11-265-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The minor occurrence of anti-addition is likely due to the geometrical isomerization of the alkenylcopper species (D/D') through a copper(I) allenoate complex (E, Figure 2) [15,26]. The resulting allenoate E can undergo protonolysis to form either syn-4 or anti-4 depending on the substituent effects of R1 and R2, while the isomerized alkenylcopper(I) D' should preferentially yield anti-4. The reduction of syn selectivity in the reaction with PPh3, PCy3 and DPPE may also be due to this isomerization (Table 1, entries 2–4).
![[1860-5397-11-265-2]](/bjoc/content/figures/1860-5397-11-265-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Isomerization of the alkenylcopper intermediates.
Figure 2: Isomerization of the alkenylcopper intermediates.
Conclusion
In summary, a copper-catalyzed conjugate addition of alkylboranes (alkyl-9-BBN) to alkynoates to form β-disubstituted acrylates is reported. The addition occurred in a formal syn-hydroalkylation mode. The stereoselectivity was excellent regardless of the substrate structure. The availability of alkylboranes through in situ alkene hydroboration is an attractive feature of this protocol and various functional groups are tolerated in both the alkylborane and alkynoate substrates.
Experimental
The reaction shown in Scheme 1 was conducted in a similar manner as described before [15]. Styrene (1a, 33 μL, 0.289 mmol) and (9-BBN-H)2 (33.6 mg, 0.138 mmol) were placed in a vial containing a magnetic stirring bar. The vial was sealed with a Teflon®-coated silicon rubber septum, and the vial was evacuated and filled with argon. 1,4-Dioxane (0.4 mL) was added to the vial, and the mixture was stirred at 60 °C for 1 h to prepare an alkylborane 2a. Meanwhile, CuOAc (1.5 mg, 0.0125 mmol), P(OPh)3 (6.9 μL, 0.025 mmol) and t-BuOK (1.4 mg, 0.0125 mmol) were placed in another vial. The vial was sealed with a Teflon®-coated silicon rubber septum, evacuated, and then filled with argon. After 1,4-dioxane (0.6 mL) was added to the vial, the mixture was stirred at 25 °C for 1 h. Next, the alkylborane solution was transferred to the vial containing the Cu(I) complex, followed by the addition of alkynoate 3a (41.3 μL, 0.25 mmol) and t-BuOH (24 μL, 0.25 mmol). After 12 h stirring at 40 °C, diethyl ether was added to the mixture. The mixture was filtered through a short plug of silica gel, which was then washed with diethyl ether. After the solvent was removed under reduced pressure, flash chromatography on silica gel (0–5% EtOAc/hexane) provided 4aa (69.4 mg, 0.248 mmol) in 99% yield with >99:1 syn/anti selectivity.
Supporting Information
| Supporting Information File 1: Experimental procedures, spectroscopic and analytical data, and copies of NMR spectra for newly synthesized compounds. | ||
| Format: PDF | Size: 5.5 MB | Download |
References
-
Corey, E. J.; Katzenellenbogen, J. A. J. Am. Chem. Soc. 1969, 91, 1851–1852. doi:10.1021/ja01035a045
Return to citation in text: [1] -
Siddall, J. B.; Biskup, M.; Fried, J. H. J. Am. Chem. Soc. 1969, 91, 1853–1854. doi:10.1021/ja01035a046
Return to citation in text: [1] -
Klein, J.; Turkel, R. M. J. Am. Chem. Soc. 1969, 91, 6186–6187. doi:10.1021/ja01050a047
Return to citation in text: [1] -
Li, G.; Wei, H.-X.; Whittlesey, B. R.; Batrice, N. N. J. Org. Chem. 1999, 64, 1061–1064. doi:10.1021/jo981976l
Return to citation in text: [1] -
Yeh, M. C. P.; Knochel, P. Tetrahedron Lett. 1989, 30, 4799–4802. doi:10.1016/S0040-4039(01)80511-6
Return to citation in text: [1] -
Yamamoto, Y.; Yatagai, H.; Maruyama, K. J. Org. Chem. 1979, 44, 1744–1746. doi:10.1021/jo01324a047
Return to citation in text: [1] -
Mueller, A. J.; Jennings, M. P. Org. Lett. 2007, 9, 5327–5329. doi:10.1021/ol702546w
Return to citation in text: [1] -
Jennings, M. P.; Sawant, K. B. Eur. J. Org. Chem. 2004, 3201–3204. doi:10.1002/ejoc.200400314
Return to citation in text: [1] -
Yamamoto, Y.; Kirai, N.; Harada, Y. Chem. Commun. 2008, 2010–2012. doi:10.1039/b802231c
Return to citation in text: [1] -
Yamamoto, Y.; Yamada, S.; Nishiyama, H. Chem. – Eur. J. 2012, 18, 3153–3156. doi:10.1002/chem.201103697
Return to citation in text: [1] -
Rajagopal, T.; Ogilvie, W. W. Synlett 2011, 1113–1116. doi:10.1055/s-0030-1259933
Return to citation in text: [1] -
Ohmiya, H.; Yoshida, M.; Sawamura, M. Org. Lett. 2011, 13, 482–485. doi:10.1021/ol102819k
Return to citation in text: [1] [2] -
Yoshida, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 11896–11899. doi:10.1021/ja304481a
Return to citation in text: [1] [2] -
Ohmiya, H.; Shido, Y.; Yoshida, M.; Sawamura, M. Chem. Lett. 2011, 40, 928–930. doi:10.1246/cl.2011.928
Return to citation in text: [1] [2] -
Wakamatsu, T.; Nagao, K.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2013, 52, 11620–11623. doi:10.1002/anie.201305973
Return to citation in text: [1] [2] [3] [4] -
Hayashi, T.; Inoue, K.; Taniguchi, N.; Ogasawara, M. J. Am. Chem. Soc. 2001, 123, 9918–9919. doi:10.1021/ja0165234
Return to citation in text: [1] -
Oh, C. H.; Jung, H. H.; Kim, K. S.; Kim, N. Angew. Chem., Int. Ed. 2003, 42, 805–808. doi:10.1002/anie.200390214
Return to citation in text: [1] -
Lin, P.-S.; Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 11296–11299. doi:10.1002/chem.200801858
Return to citation in text: [1] -
Bush, A. G.; Jiang, J. L.; Payne, P. R.; Ogilvie, W. W. Tetrahedron 2009, 65, 8502–8506. doi:10.1016/j.tet.2009.08.029
Return to citation in text: [1] -
Ohmiya, H.; Yokobori, U.; Makida, Y.; Sawamura, M. J. Am. Chem. Soc. 2010, 132, 2895–2897. doi:10.1021/ja9109105
Return to citation in text: [1] -
Nagao, K.; Ohmiya, H.; Sawamura, M. Synthesis 2012, 44, 1535–1541. doi:10.1055/s-0031-1290818
Return to citation in text: [1] -
Nagao, K.; Yokobori, U.; Makida, Y.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 8982–8987. doi:10.1021/ja302520h
Return to citation in text: [1] -
Ohmiya, H.; Yokobori, U.; Makida, Y.; Sawamura, M. Org. Lett. 2011, 13, 6312–6315. doi:10.1021/ol202866h
Return to citation in text: [1] -
Shido, Y.; Yoshida, M.; Tanabe, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 18573–18576. doi:10.1021/ja3093955
Return to citation in text: [1] -
Hojoh, K.; Shido, Y.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2014, 53, 4954–4958. doi:10.1002/anie.201402386
Return to citation in text: [1] -
Nilsson, K.; Andersson, T.; Ullenius, C.; Gerold, A.; Krause, N. Chem. – Eur. J. 1998, 4, 2051–2058. doi:10.1002/(SICI)1521-3765(19981002)4:10<2051::AID-CHEM2051>3.0.CO;2-F
Return to citation in text: [1]
| 1. | Corey, E. J.; Katzenellenbogen, J. A. J. Am. Chem. Soc. 1969, 91, 1851–1852. doi:10.1021/ja01035a045 |
| 2. | Siddall, J. B.; Biskup, M.; Fried, J. H. J. Am. Chem. Soc. 1969, 91, 1853–1854. doi:10.1021/ja01035a046 |
| 3. | Klein, J.; Turkel, R. M. J. Am. Chem. Soc. 1969, 91, 6186–6187. doi:10.1021/ja01050a047 |
| 4. | Li, G.; Wei, H.-X.; Whittlesey, B. R.; Batrice, N. N. J. Org. Chem. 1999, 64, 1061–1064. doi:10.1021/jo981976l |
| 5. | Yeh, M. C. P.; Knochel, P. Tetrahedron Lett. 1989, 30, 4799–4802. doi:10.1016/S0040-4039(01)80511-6 |
| 6. | Yamamoto, Y.; Yatagai, H.; Maruyama, K. J. Org. Chem. 1979, 44, 1744–1746. doi:10.1021/jo01324a047 |
| 7. | Mueller, A. J.; Jennings, M. P. Org. Lett. 2007, 9, 5327–5329. doi:10.1021/ol702546w |
| 8. | Jennings, M. P.; Sawant, K. B. Eur. J. Org. Chem. 2004, 3201–3204. doi:10.1002/ejoc.200400314 |
| 15. | Wakamatsu, T.; Nagao, K.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2013, 52, 11620–11623. doi:10.1002/anie.201305973 |
| 12. | Ohmiya, H.; Yoshida, M.; Sawamura, M. Org. Lett. 2011, 13, 482–485. doi:10.1021/ol102819k |
| 13. | Yoshida, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 11896–11899. doi:10.1021/ja304481a |
| 14. | Ohmiya, H.; Shido, Y.; Yoshida, M.; Sawamura, M. Chem. Lett. 2011, 40, 928–930. doi:10.1246/cl.2011.928 |
| 11. | Rajagopal, T.; Ogilvie, W. W. Synlett 2011, 1113–1116. doi:10.1055/s-0030-1259933 |
| 9. | Yamamoto, Y.; Kirai, N.; Harada, Y. Chem. Commun. 2008, 2010–2012. doi:10.1039/b802231c |
| 10. | Yamamoto, Y.; Yamada, S.; Nishiyama, H. Chem. – Eur. J. 2012, 18, 3153–3156. doi:10.1002/chem.201103697 |
| 15. | Wakamatsu, T.; Nagao, K.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2013, 52, 11620–11623. doi:10.1002/anie.201305973 |
| 15. | Wakamatsu, T.; Nagao, K.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2013, 52, 11620–11623. doi:10.1002/anie.201305973 |
| 26. | Nilsson, K.; Andersson, T.; Ullenius, C.; Gerold, A.; Krause, N. Chem. – Eur. J. 1998, 4, 2051–2058. doi:10.1002/(SICI)1521-3765(19981002)4:10<2051::AID-CHEM2051>3.0.CO;2-F |
| 12. | Ohmiya, H.; Yoshida, M.; Sawamura, M. Org. Lett. 2011, 13, 482–485. doi:10.1021/ol102819k |
| 13. | Yoshida, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 11896–11899. doi:10.1021/ja304481a |
| 14. | Ohmiya, H.; Shido, Y.; Yoshida, M.; Sawamura, M. Chem. Lett. 2011, 40, 928–930. doi:10.1246/cl.2011.928 |
| 15. | Wakamatsu, T.; Nagao, K.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2013, 52, 11620–11623. doi:10.1002/anie.201305973 |
| 20. | Ohmiya, H.; Yokobori, U.; Makida, Y.; Sawamura, M. J. Am. Chem. Soc. 2010, 132, 2895–2897. doi:10.1021/ja9109105 |
| 21. | Nagao, K.; Ohmiya, H.; Sawamura, M. Synthesis 2012, 44, 1535–1541. doi:10.1055/s-0031-1290818 |
| 22. | Nagao, K.; Yokobori, U.; Makida, Y.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 8982–8987. doi:10.1021/ja302520h |
| 23. | Ohmiya, H.; Yokobori, U.; Makida, Y.; Sawamura, M. Org. Lett. 2011, 13, 6312–6315. doi:10.1021/ol202866h |
| 24. | Shido, Y.; Yoshida, M.; Tanabe, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 18573–18576. doi:10.1021/ja3093955 |
| 25. | Hojoh, K.; Shido, Y.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2014, 53, 4954–4958. doi:10.1002/anie.201402386 |
| 16. | Hayashi, T.; Inoue, K.; Taniguchi, N.; Ogasawara, M. J. Am. Chem. Soc. 2001, 123, 9918–9919. doi:10.1021/ja0165234 |
| 17. | Oh, C. H.; Jung, H. H.; Kim, K. S.; Kim, N. Angew. Chem., Int. Ed. 2003, 42, 805–808. doi:10.1002/anie.200390214 |
| 18. | Lin, P.-S.; Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 11296–11299. doi:10.1002/chem.200801858 |
| 19. | Bush, A. G.; Jiang, J. L.; Payne, P. R.; Ogilvie, W. W. Tetrahedron 2009, 65, 8502–8506. doi:10.1016/j.tet.2009.08.029 |
© 2015 Wakamatsu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)