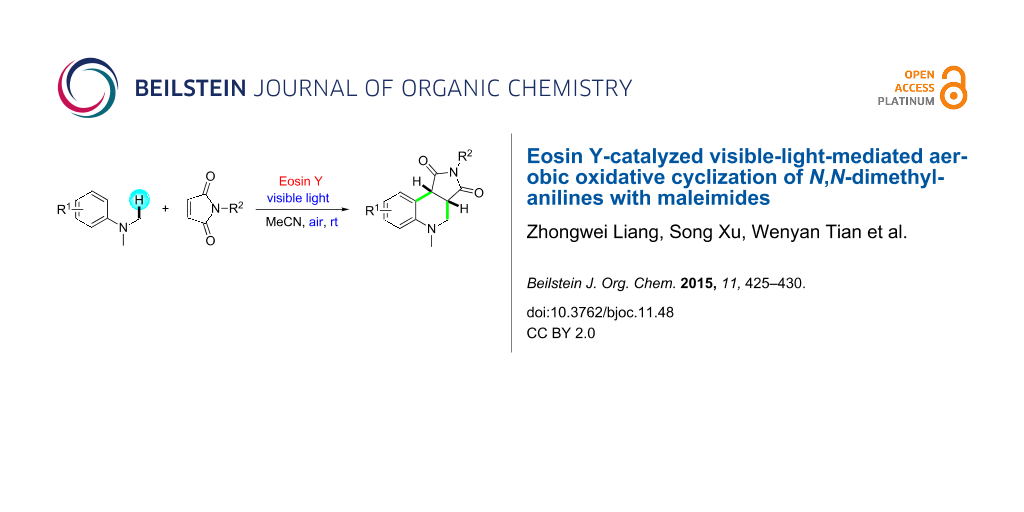
Abstract
A novel and simple strategy for the efficient synthesis of the corresponding tetrahydroquinolines from N,N-dimethylanilines and maleimides using visible light in an air atmosphere in the presence of Eosin Y as a photocatalyst has been developed. The metal-free protocol involves aerobic oxidative cyclization via sp3 C–H bond functionalization process to afford good yields in a one-pot procedure under mild conditions.

Graphical Abstract
Introduction
Over the past several years, visible light photoredox catalysis has become a powerful and promising tool and has been productively used to drive chemical transformations in the field of organic synthesis [1-6]. The approach takes full advantage of visible light, which is clean, abundant, and renewable. The pioneering work in this research area, reported by the groups of MacMillan [7-9], Yoon [10,11], Stephenson [12,13] and others [14-18], has demonstrated that ruthenium and iridium complexes as visible light photoredox catalysts are capable of catalyzing a broad range of useful reactions. A variety of new methods have been developed to accomplish known and new chemical transformations by means of these transition metal-based photocatalysts so far.
However, the ruthenium and iridium catalysts usually are high-cost, potentially toxic and not sustainable. Similar to the redox properties of these organometallic complexes, some metal-free organic dyes such as Eosin Y, Rose Bengal, Fluorescein, and Methylene Blue, have shown superiority of their applications as photocatalysts, which are easy to handle, environmentally friendly, inexpensive, and have great potential for applications in visible-light-mediated photoredox reactions [19-27].
More recently, visible-light-induced sp3 C–H bond functionalization adjacent to nitrogen atoms has been extensively studied and has become a fundamental organic transformation [28-38]. Tertiary amine A generally generates a nucleophilic α-aminoalkyl radical B or an electrophilic iminium ion C via visible-light photoredox catalysis. Unfortunately the research of the α-aminoalkyl radical is limited in photochemical synthesis because it tends to form the iminium ion by one electron oxidation (Scheme 1) [39-41].
![[1860-5397-11-48-i1]](/bjoc/content/inline/1860-5397-11-48-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Visible-light-induced sp3 C–H bond functionalization of tertiary amines.
Scheme 1: Visible-light-induced sp3 C–H bond functionalization of tertiary amines.
In the context of this research background, we investigated the α-aminoalkyl radical route to achieve the aerobic oxidative cyclization of N,N-dimethylanilines with maleimides to form the corresponding tetrahydroquinoline derivatives under organic dye Eosin Y catalysis. Swan and Roy reported the reaction using benzoyl peroxide as catalyst at low temperature as early as 1968 [42]. In 2011, Miura and co-workers achieved this transformation using a copper catalyst and air as the terminal oxidant [43]. Bian and co-workers presented the same reaction using [Ru(bpy)3]3+ as photoredox catalyst under irradiation with visible light next year [44]. Herein, we show an environmentally friendly aerobic oxidative cyclization methodology that avoids the use of metal catalysts and makes full use of air as oxidant.
Results and Discussion
Our investigations for the envisaged protocol commenced with the reaction of N,N-dimethylaniline (1a) (0.5 mmol) with N-phenylmaleimide (2a) (0.25 mmol) in MeCN (3 mL) in the presence of 3 mol % Eosin Y under an air atmosphere (with no air bubbling). The reaction mixture was irradiated with visible light (two 9 W blue LEDs) at room temperature. Gratifyingly, the desired product tetrahydroquinoline 3a was obtained in 82% yield after 18 h (Table 1, entry 1). We screened a number of metal-free organic dyes for photocatalysts. Of the dyes screened, Eosin Y showed the highest efficiency (Table 1, entry 1), Rose Bengal gave a slightly lower yield (Table 1, entry 2), whereas Methylene Blue and Fluorescein gave poor yields (Table 1, entries 3 and 4). Under an O2 atmosphere, the yield of tetrahydroquinoline product 3a was decreased to 77% (Table 1, entry 5). When the molar proportion of 1a and 2a was adjusted to 1:1 and 1:2, the yield of 3a decreased (Table 1, entries 6 and 7). Then, a series of control experiments were carried out, which indicated that Eosin Y, visible light and air are all essential for the reaction (Table 1, entries 8–10).
Table 1: Screening and control experimentsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-48-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Organic dye | Oxidant | Yield (%)b |
|---|---|---|---|
| 1 | Eosin Y | air | 82 |
| 2 | Rose Bengal | air | 67 |
| 3 | Methylene Blue | air | trace |
| 4 | Fluorescein | air | trace |
| 5 | Eosin Y | O2c | 77 |
| 6 | Eosin Y | air | 69d |
| 7 | Eosin Y | air | 73e |
| 8 | none | air | n.r. |
| 9 | Eosin Y | air | n.r.f |
| 10 | Eosin Y | none | traceg |
aReaction conditions: 1a (0.5 mmol), 2a (0.25 mmol), organic dye (3 mol %), MeCN (3 mL), two 9 W Blue LEDs irradiation under an air atmosphere at rt. bIsolated yield of the product 3a; n.r. = no reaction. cUnder O2 (1 atm, balloon). d0.25 mmol of 1a and 0.25 mmol of 2a were used. e0.25 mmol of 1a and 0.5 mmol of 2a were used. fThe reaction was carried out in the dark. gUnder N2.
Next, we optimized the reaction conditions with respect to solvent and catalyst dosage. MeCN was found to be the best solvent (Table 2, entry 1) among DMF, DCE, DCM, DMSO, acetone, dioxane, and MeNO2. When the amount of Eosin Y was decreased from 3 mol % to 2 mol % or increased from 3 mol % to 4 mol %, the yield of the tetrahydroquinoline product 3a was slightly reduced (Table 2, entries 9 and 10). Thus, the optimum catalyst dosage of Eosin Y was found to be 3 mol %.
Table 2: Optimization of reaction conditionsa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-48-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Eosin Y (mol %) | Solvent | Yield (%)b |
|---|---|---|---|
| 1 | 3 | MeCN | 82 |
| 2 | 3 | DMF | 47 |
| 3 | 3 | DCE | 71 |
| 4 | 3 | DCM | 37 |
| 5 | 3 | DMSO | trace |
| 6 | 3 | acetone | 64 |
| 7 | 3 | dioxane | 51 |
| 8 | 3 | MeNO2 | 58 |
| 9 | 2 | MeCN | 80 |
| 10 | 4 | MeCN | 77 |
aReaction conditions: 1a (0.5 mmol), 2a (0.25 mmol), solvent (3 mL), two 9 W blue LEDs irradiation under an air atmosphere at rt. bIsolated yield of the product 3a.
With the optimized conditions in hand, the substrate scope of this reaction was examined (Scheme 2). The reaction is mild and tolerates many functional groups. N,N-dimethylaniline and substituted N,N-dimethylanilines incorporating methyl and bromo on the phenyl ring reacted with 2 to afford the corresponding tetrahydroquinolines 3 in good yields. N-arylmaleimides with electron-donating groups such as methyl, methoxy and electron-withdrawing groups such as chloro, bromo, and N-methylmaleimide all underwent the aerobic oxidative cyclization to give the corresponding products in good yields. When using 4,4'-methylenebis(N,N-dimethylaniline) as the substrate, the reaction occurred only on one side and the yield of the product 3p is 52%. The reaction of N,N,3-trimethylaniline and N-phenylmaleimide resulted in the formation of a mixture of regioisomers 3q1 and 3q2 with 81% combined yield. The major product was the sterically more hindered 3q1 [43-45].
![[1860-5397-11-48-i2]](/bjoc/content/inline/1860-5397-11-48-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope for aerobic oxidative cyclization of N,N-dimethylanilines with maleimides.
Scheme 2: Substrate scope for aerobic oxidative cyclization of N,N-dimethylanilines with maleimides.
On the basis of our observations and literature reported [19,36,38,43,44], a proposed mechanism for the formation of the corresponding tetrahydroquinolines 3 form N,N-dimethylanilines 1 and maleimides 2 is depicted in Scheme 3. On absorption of visible light, the ground state of Eosin Y (EY) is induced to its single excited state (1EY*), which moves to its more stable triplet excited state (3EY*) through inter system crossing (ISC) [46,47]. 3EY* may undergo an oxidative or reductive quenching cycle [48-50]. In this mechanism, a single electron transfer (SET) from 1 to 3EY* generates the amine radical cation 4, and at the same time, 3EY* is reduced to the EY•−. In the presence of oxygen, the photoredox catalytic cycle of EY is finished via a SET oxidation, with the production of a superoxide radical anion O2•−. Deprotonation of 4 generates α-aminoalkyl radical 5. Then 5 reacts with 2 to generate radical 6, and the latter then undergoes cyclization to form intermediate 7. Proton and electron transfer from 7 to O2•− yields the final product 3 and HOO−. The HOO− will be subsequently protonated to yield H2O2 as the by-product. H2O2 was detected after the reaction was completed by using KI/starch indicator (see the Supporting Information File 1). The involvement of radical pathway was supported by experimental result that the reaction was suppressed in the presence of TEMPO.
![[1860-5397-11-48-i3]](/bjoc/content/inline/1860-5397-11-48-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we report an efficient metal-free method for the synthesis of corresponding tetrahydroquinolines from N,N-dimethylanilines and maleimides using molecular oxygen as oxidant and Eosin Y as catalyst under the irradiation of visible light. The protocol is significantly green because it utilizes visible light and atmospheric oxygen as the greenest reagents, and metal-free, cheap Eosin Y with a relatively low loading as the photocatalyst to deliver the product at room temperature in a simple one-pot procedure. This methodology expands the range of substrates in the area of visible light photoredox reactions.
Supporting Information
| Supporting Information File 1: Experimental section and characterization of the synthesized compounds. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223
Return to citation in text: [1] -
Schultz, D. M.; Yoon, T. P. Science 2014, 343, 985–993. doi:10.1126/science.1239176
Return to citation in text: [1] -
Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056
Return to citation in text: [1] -
Pirnot, M. T.; Rankic, D. A.; Martin, D. B. C.; MacMillan, D. W. C. Science 2013, 339, 1593–1596. doi:10.1126/science.1232993
Return to citation in text: [1] -
Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77–80. doi:10.1126/science.1161976
Return to citation in text: [1] -
Nagib, D. A.; MacMillan, D. W. C. Nature 2011, 480, 224–228. doi:10.1038/nature10647
Return to citation in text: [1] -
Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. doi:10.1021/ja805387f
Return to citation in text: [1] -
Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2009, 131, 14604–14605. doi:10.1021/ja903732v
Return to citation in text: [1] -
Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949
Return to citation in text: [1] -
Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582
Return to citation in text: [1] -
Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2011, 50, 7171–7175. doi:10.1002/anie.201102306
Return to citation in text: [1] -
Zou, Y.-Q.; Chen, J.-R.; Liu, X.-P.; Lu, L.-Q.; Davis, R.-L.; Jørgensen, K. A.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 784–788. doi:10.1002/anie.201107028
Return to citation in text: [1] -
Fan, W.; Li, P. Angew. Chem., Int. Ed. 2014, 53, 12201–12204. doi:10.1002/anie.201407413
Return to citation in text: [1] -
Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D. P.; Rueping, M. J. Am. Chem. Soc. 2013, 135, 1823–1829. doi:10.1021/ja309580a
Return to citation in text: [1] -
Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Org. Lett. 2013, 15, 624–627. doi:10.1021/ol303437m
Return to citation in text: [1] -
Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951–954. doi:10.1002/anie.201002992
Return to citation in text: [1] [2] -
Liu, H.; Feng, W.; Kee, C. W.; Zhao, Y.; Leow, D.; Pan, Y.; Tan, C.-H. Green Chem. 2010, 12, 953–956. doi:10.1039/b924609f
Return to citation in text: [1] -
Gu, X.; Li, X.; Chai, Y.; Yang, Q.; Li, P.; Yao, Y. Green Chem. 2013, 15, 357–361. doi:10.1039/C2GC36683E
Return to citation in text: [1] -
Li, X.; Gu, X.; Li, Y.; Li, P. ACS Catal. 2014, 4, 1897–1900. doi:10.1021/cs5005129
Return to citation in text: [1] -
Pitre, S. P.; McTiernan, C. D.; Ismaili, H.; Scaiano, J. C. J. Am. Chem. Soc. 2013, 135, 13286–13289. doi:10.1021/ja406311g
Return to citation in text: [1] -
Vila, C.; Lau, J.; Rueping, M. Beilstein J. Org. Chem. 2014, 10, 1233–1238. doi:10.3762/bjoc.10.122
Return to citation in text: [1] -
Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852–3855. doi:10.1021/ol201376v
Return to citation in text: [1] -
Chen, L.; Chao, C. S.; Pan, Y.; Dong, S.; Teo, Y. C.; Wang, J.; Tan, C.-H. Org. Biomol. Chem. 2013, 11, 5922–5925. doi:10.1039/c3ob41091a
Return to citation in text: [1] -
Majek, M.; Filace, F.; Jacobi von Wangelin, A. Beilstein J. Org. Chem. 2014, 10, 981–989. doi:10.3762/bjoc.10.97
Return to citation in text: [1] -
Dai, X.; Cheng, D.; Guan, B.; Mao, W.; Xu, X.; Li, X. J. Org. Chem. 2014, 79, 7212–7219. doi:10.1021/jo501097b
Return to citation in text: [1] -
Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j
Return to citation in text: [1] -
Rueping, M.; Leonori, D.; Poisson, T. Chem. Commun. 2011, 47, 9615–9617. doi:10.1039/c1cc13660g
Return to citation in text: [1] -
Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 12709–12711. doi:10.1039/c1cc15643h
Return to citation in text: [1] -
McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920
Return to citation in text: [1] -
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Chem. – Eur. J. 2012, 18, 16473–16477. doi:10.1002/chem.201203066
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050
Return to citation in text: [1] -
Zhu, S.; Rueping, M. Chem. Commun. 2012, 48, 11960–11962. doi:10.1039/c2cc36995h
Return to citation in text: [1] -
Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234
Return to citation in text: [1] [2] -
Ruiz Espelt, L.; Wiensch, E. M.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107–4114. doi:10.1021/jo400428m
Return to citation in text: [1] -
Xie, J.; Jin, H.; Xu, P.; Zhu, C. Tetrahedron Lett. 2014, 55, 36–48. doi:10.1016/j.tetlet.2013.10.090
Return to citation in text: [1] [2] -
Wayner, D. D. M.; Dannenberg, J. J.; Griller, D. Chem. Phys. Lett. 1986, 131, 189–191. doi:10.1016/0009-2614(86)80542-5
Return to citation in text: [1] -
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y
Return to citation in text: [1] -
Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t
Return to citation in text: [1] -
Roy, R. B.; Swan, G. A. Chem. Commun. 1968, 1446–1447. doi:10.1039/C19680001446
Return to citation in text: [1] -
Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2011, 76, 6447–6451. doi:10.1021/jo2011329
Return to citation in text: [1] [2] [3] -
Ju, X.; Li, D.; Li, W.; Yu, W.; Bian, F. Adv. Synth. Catal. 2012, 354, 3561–3567. doi:10.1002/adsc.201200608
Return to citation in text: [1] [2] [3] -
Ju, X.; Liang, Y.; Jia, P.; Li, W.; Yu, W. Org. Biomol. Chem. 2012, 10, 498–501. doi:10.1039/c1ob06652h
Return to citation in text: [1] -
Shimidzu, T.; Iyoda, T.; Koide, Y. J. Am. Chem. Soc. 1985, 107, 35–41. doi:10.1021/ja00287a007
Return to citation in text: [1] -
Neckers, D. C.; Valdes-Aguilera, O. M. Photochemistry of the Xanthene Dyes. In Adv. Photochem.; Volman, D. H.; Hammond, G. S.; Neckers, D. C., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1993; Vol. 18, pp 315–394. doi:10.1002/9780470133491.ch4
Return to citation in text: [1] -
Lazarides, T.; McCormick, T.; Du, P.; Luo, G.; Lindley, B.; Eisenberg, R. J. Am. Chem. Soc. 2009, 131, 9192–9194. doi:10.1021/ja903044n
Return to citation in text: [1] -
Lee, S. H.; Nam, D. H.; Park, C. B. Adv. Synth. Catal. 2009, 351, 2589–2594. doi:10.1002/adsc.200900547
Return to citation in text: [1] -
Encinas, M. V.; Rufs, A. M.; Bertolotti, S. G.; Previtali, C. M. Polymer 2009, 50, 2762–2767. doi:10.1016/j.polymer.2009.04.024
Return to citation in text: [1]
| 1. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 2. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 3. | Schultz, D. M.; Yoon, T. P. Science 2014, 343, 985–993. doi:10.1126/science.1239176 |
| 4. | Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687 |
| 5. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 6. | Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056 |
| 14. | Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2011, 50, 7171–7175. doi:10.1002/anie.201102306 |
| 15. | Zou, Y.-Q.; Chen, J.-R.; Liu, X.-P.; Lu, L.-Q.; Davis, R.-L.; Jørgensen, K. A.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 784–788. doi:10.1002/anie.201107028 |
| 16. | Fan, W.; Li, P. Angew. Chem., Int. Ed. 2014, 53, 12201–12204. doi:10.1002/anie.201407413 |
| 17. | Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D. P.; Rueping, M. J. Am. Chem. Soc. 2013, 135, 1823–1829. doi:10.1021/ja309580a |
| 18. | Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Org. Lett. 2013, 15, 624–627. doi:10.1021/ol303437m |
| 48. | Lazarides, T.; McCormick, T.; Du, P.; Luo, G.; Lindley, B.; Eisenberg, R. J. Am. Chem. Soc. 2009, 131, 9192–9194. doi:10.1021/ja903044n |
| 49. | Lee, S. H.; Nam, D. H.; Park, C. B. Adv. Synth. Catal. 2009, 351, 2589–2594. doi:10.1002/adsc.200900547 |
| 50. | Encinas, M. V.; Rufs, A. M.; Bertolotti, S. G.; Previtali, C. M. Polymer 2009, 50, 2762–2767. doi:10.1016/j.polymer.2009.04.024 |
| 12. | Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949 |
| 13. | Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582 |
| 10. | Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. doi:10.1021/ja805387f |
| 11. | Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2009, 131, 14604–14605. doi:10.1021/ja903732v |
| 19. | Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951–954. doi:10.1002/anie.201002992 |
| 36. | Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234 |
| 38. | Xie, J.; Jin, H.; Xu, P.; Zhu, C. Tetrahedron Lett. 2014, 55, 36–48. doi:10.1016/j.tetlet.2013.10.090 |
| 43. | Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2011, 76, 6447–6451. doi:10.1021/jo2011329 |
| 44. | Ju, X.; Li, D.; Li, W.; Yu, W.; Bian, F. Adv. Synth. Catal. 2012, 354, 3561–3567. doi:10.1002/adsc.201200608 |
| 7. | Pirnot, M. T.; Rankic, D. A.; Martin, D. B. C.; MacMillan, D. W. C. Science 2013, 339, 1593–1596. doi:10.1126/science.1232993 |
| 8. | Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77–80. doi:10.1126/science.1161976 |
| 9. | Nagib, D. A.; MacMillan, D. W. C. Nature 2011, 480, 224–228. doi:10.1038/nature10647 |
| 46. | Shimidzu, T.; Iyoda, T.; Koide, Y. J. Am. Chem. Soc. 1985, 107, 35–41. doi:10.1021/ja00287a007 |
| 47. | Neckers, D. C.; Valdes-Aguilera, O. M. Photochemistry of the Xanthene Dyes. In Adv. Photochem.; Volman, D. H.; Hammond, G. S.; Neckers, D. C., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1993; Vol. 18, pp 315–394. doi:10.1002/9780470133491.ch4 |
| 42. | Roy, R. B.; Swan, G. A. Chem. Commun. 1968, 1446–1447. doi:10.1039/C19680001446 |
| 44. | Ju, X.; Li, D.; Li, W.; Yu, W.; Bian, F. Adv. Synth. Catal. 2012, 354, 3561–3567. doi:10.1002/adsc.201200608 |
| 39. | Wayner, D. D. M.; Dannenberg, J. J.; Griller, D. Chem. Phys. Lett. 1986, 131, 189–191. doi:10.1016/0009-2614(86)80542-5 |
| 40. | Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y |
| 41. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 43. | Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2011, 76, 6447–6451. doi:10.1021/jo2011329 |
| 44. | Ju, X.; Li, D.; Li, W.; Yu, W.; Bian, F. Adv. Synth. Catal. 2012, 354, 3561–3567. doi:10.1002/adsc.201200608 |
| 45. | Ju, X.; Liang, Y.; Jia, P.; Li, W.; Yu, W. Org. Biomol. Chem. 2012, 10, 498–501. doi:10.1039/c1ob06652h |
| 28. | Dai, X.; Cheng, D.; Guan, B.; Mao, W.; Xu, X.; Li, X. J. Org. Chem. 2014, 79, 7212–7219. doi:10.1021/jo501097b |
| 29. | Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j |
| 30. | Rueping, M.; Leonori, D.; Poisson, T. Chem. Commun. 2011, 47, 9615–9617. doi:10.1039/c1cc13660g |
| 31. | Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 12709–12711. doi:10.1039/c1cc15643h |
| 32. | McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920 |
| 33. | Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Chem. – Eur. J. 2012, 18, 16473–16477. doi:10.1002/chem.201203066 |
| 34. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 35. | Zhu, S.; Rueping, M. Chem. Commun. 2012, 48, 11960–11962. doi:10.1039/c2cc36995h |
| 36. | Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234 |
| 37. | Ruiz Espelt, L.; Wiensch, E. M.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107–4114. doi:10.1021/jo400428m |
| 38. | Xie, J.; Jin, H.; Xu, P.; Zhu, C. Tetrahedron Lett. 2014, 55, 36–48. doi:10.1016/j.tetlet.2013.10.090 |
| 19. | Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951–954. doi:10.1002/anie.201002992 |
| 20. | Liu, H.; Feng, W.; Kee, C. W.; Zhao, Y.; Leow, D.; Pan, Y.; Tan, C.-H. Green Chem. 2010, 12, 953–956. doi:10.1039/b924609f |
| 21. | Gu, X.; Li, X.; Chai, Y.; Yang, Q.; Li, P.; Yao, Y. Green Chem. 2013, 15, 357–361. doi:10.1039/C2GC36683E |
| 22. | Li, X.; Gu, X.; Li, Y.; Li, P. ACS Catal. 2014, 4, 1897–1900. doi:10.1021/cs5005129 |
| 23. | Pitre, S. P.; McTiernan, C. D.; Ismaili, H.; Scaiano, J. C. J. Am. Chem. Soc. 2013, 135, 13286–13289. doi:10.1021/ja406311g |
| 24. | Vila, C.; Lau, J.; Rueping, M. Beilstein J. Org. Chem. 2014, 10, 1233–1238. doi:10.3762/bjoc.10.122 |
| 25. | Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852–3855. doi:10.1021/ol201376v |
| 26. | Chen, L.; Chao, C. S.; Pan, Y.; Dong, S.; Teo, Y. C.; Wang, J.; Tan, C.-H. Org. Biomol. Chem. 2013, 11, 5922–5925. doi:10.1039/c3ob41091a |
| 27. | Majek, M.; Filace, F.; Jacobi von Wangelin, A. Beilstein J. Org. Chem. 2014, 10, 981–989. doi:10.3762/bjoc.10.97 |
| 43. | Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2011, 76, 6447–6451. doi:10.1021/jo2011329 |
© 2015 Liang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)