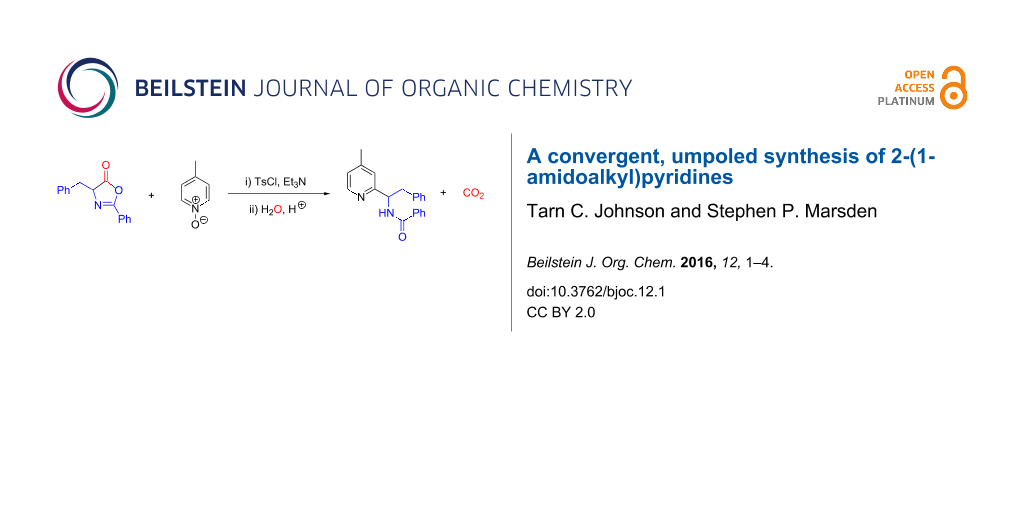
Abstract
A convenient, one-pot, two-component synthesis of 2-(1-amidoalkyl)pyridines is reported, based upon the substitution of suitably-activated pyridine N-oxides by azlactone nucleophiles, followed by decarboxylative azlactone ring-opening. The synthesis obviates the need for precious metal catalysts to achieve a formal enolate arylation reaction, and constitutes a formally ‘umpoled’ approach to this valuable class of bioactive structures.

Graphical Abstract
Introduction
Pyridines constitute the most frequently observed class of heterocycles found in pharmaceutical products [1]. As such, there is significant demand for synthetic methods that enable access both to structurally novel and diverse substituted pyridines for new medicines discovery, and for the development of clean, efficient and robust methods for their manufacture on large scale. Pyridines bearing a 1-amidoalkyl substituent at the 2-position are found in numerous biologically active natural products such as the antitumour antibiotic kedarcidin chromophore 1 [2] and the RNA polymerase inhibitor cyclothiazomycin B1 2 [3] (Figure 1). Additionally, the motif is commonly incorporated into synthetic pharmaceutical candidates, for example in the factor XIa inhibitor 3 [4], the orally-active renin inhibitor 4 [5] and the threonine tyrosine kinase inhibitor CFI-401870 (5) [6].
![[1860-5397-12-1-1]](/bjoc/content/figures/1860-5397-12-1-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of naturally-occurring and synthetic bioactive (amidoalkyl)pyridines.
Figure 1: Examples of naturally-occurring and synthetic bioactive (amidoalkyl)pyridines.
The 2-(1-amidoalkyl)pyridines are almost always synthesised by acylation of the related 2-(1-aminoalkyl)pyridines, which can be prepared by reduction of ketimines derived from 2-acylpyridines [5] or 2-cyanopyridines [6], addition of carbon nucleophiles to aldimines derived from pyridine-2-carboxaldehydes [4,7], or nucleophilic substitution of 2-(1-hydroxyalkyl)pyridines [8]. Alternatively, acyclic precursors bearing the amidoalkyl (or protected aminoalkyl) substituent can be applied in de novo construction of the pyridine ring, exemplified by approaches to the core of the cyclothiazomycins [9,10]. Regardless of the method employed, these are multistep protocols, frequently employing moisture-sensitive organometallic agents or reducing agents. In this paper, we report a new one-pot, two-component synthesis of 2-(1-amidoalkyl)pyridines that arises from a formally ‘umpoled’ coupling of an α-(amidoalkyl) anion equivalent with a pyridyl electrophile.
Results and Discussion
Reaction discovery
Our research group has a longstanding interest in the synthesis of α,α-disubstituted amino acids [11-15], and in particular has developed methods for the preparation of α-aryl variants by palladium-catalysed enolate arylation reactions [13-15]. More recently, we have sought to develop more sustainable methods for the arylation of amino acid enolate equivalents that avoid the use of precious metal salts and expensive bespoke ligands, based upon the electrophilic activation of pyridine N-oxides and subsequent reaction with acidic carbon nucleophiles [16-20]. Specifically, we have demonstrated that α-pyridyl,α-alkylamino acid derivatives can be prepared in a one-pot three component coupling between readily-available azlactones and pyridine N-oxides in the presence of p-toluenesulfonyl chloride as an activating agent, followed by opening of the arylated azlactone intermediate with nucleophiles such as alcohols, primary and secondary amines, and N,O-dialkylhydroxylamines [21] (Scheme 1). However, in the reaction of alanine-derived azlactone 6 with 4-methylpyridine N-oxide (7) we found that when water (in the form of 1 M HCl) was employed as a nucleophile, the product isolated was in fact the 2-(1-benzamidoethyl)-4-methylpyridine (8a), formed by facile decarboxylation of the intermediate pyridylamino acid 9 (Nu = OH) [22,23]. After some minor process optimisation, this product was isolated in 64% yield.
![[1860-5397-12-1-i1]](/bjoc/content/inline/1860-5397-12-1-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Discovery of the azlactone arylation/decarboxylative hydrolysis approach to 2-(1-amidoalkyl)pyridines.
Scheme 1: Discovery of the azlactone arylation/decarboxylative hydrolysis approach to 2-(1-amidoalkyl)pyridin...
We recognised that this constitutes a formally ‘umpoled’ [24] coupling of an α-amino- or amidoalkyl anion [25-27] with a pyridyl electrophile and hence would complement existing synthetic methods. Given the ready availability of azlactones bearing differential functionality at C2 and C4, a wide range of 2-(1-amidoalkyl)pyridines should become available and we elected to exemplify this process.
Substrate scope
We first examined variation of azlactone substituents in their reaction with 4-methylpyridine N-oxide (Scheme 2). Pleasingly, bulkier C4-substituents such as benzyl or isobutyl groups (derived from phenylalanine and leucine, respectively) were well tolerated, with isolated yields for the products 8b and 8c of 75% and 69%, respectively. Variation of the C2-azlactone substituent was also evaluated: swapping a phenyl substituent for a tert-butyl substituent gave rise to the pivaloyl amide 8d, albeit in a modest 36% yield.
![[1860-5397-12-1-i2]](/bjoc/content/inline/1860-5397-12-1-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the direct amidoalkylation of pyridine N-oxides.
Scheme 2: Substrate scope of the direct amidoalkylation of pyridine N-oxides.
Next we examined the behaviour of different 4-substituted pyridine N-oxides (either commercially available or prepared by oxidation of the corresponding pyridine with m-chloroperbenzoic acid) with a leucine-derived azlactone. The presence of bulkier alkyl or aromatic substituents was well tolerated, giving products 8e and 8f in good yield; however, the presence of an electron-donating methoxy group led to a lower yield of 8g (45%). This may reflect the lower electrophilicity of the activated pyridine N-oxide, which may allow non-productive azlactone decomposition pathways to compete with the desired substitution.
Finally, we examined the regiochemical outcome of the reaction of pyridine N-oxides with other substitution patterns. Pyridine N-oxide itself delivered a ca. 2:1 mixture of the separable 4- and 2-substituted isomers of 8h in overall 60% yield. Statistically corrected for the available reactive positions, this reflects an inherent 4:1 preference for reaction at the 4-position, which may in part be due to steric hindrance at the C2-positions by the activated N-oxide. The high C4-selectivity was also evident in the formation of a >5:1 selectivity for the formation of the 2,4-isomer of 8i over the 2,6-isomer when 2-methylpyridine N-oxide was used as substrate. Finally, the use of 3-methylpyridine N-oxide gave a ca. 7:2:2 mixture of 3,4-:2,3-:2,5-isomers of 8j. Although the regioselectivities are not exceptionally high, the ready chromatographic separation of the various isomers makes this a synthetically tractable approach to 4-substituted (1-amidoalkyl)pyridine derivatives.
Conclusion
In summary, we have demonstrated a new umpoled disconnection for the one-pot, two-component synthesis of 2-(1-amidoalkyl)pyridines using simple, widely-available coupling partners without the requirement for expensive or critically-available reagents and catalysts. The reactions display good generality over 10 examples (36–75% yields, average 60%). Given the medicinal relevance of the (amidoalkyl)pyridine products, and the convergent nature of the reaction, we believe that the method should find ready application in the concise synthesis of bioactive molecules.
Acknowledgements
The research for this work has received funding from the Innovative Medicines Institute joint undertaking project CHEM21 under grant agreement no. 115360, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution.
References
-
Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337–2347. doi:10.1039/b602413k
Return to citation in text: [1] -
Leet, J. E.; Schroeder, D. R.; Langley, D. R.; Colson, K. L.; Huang, S.; Klohr, S. E.; Lee, M. S.; Golik, J.; Hofstead, S. J.; Doyle, T. W.; Matson, J. A. J. Am. Chem. Soc. 1993, 115, 8432–8443. doi:10.1021/ja00071a062
Return to citation in text: [1] -
Hashimoto, M.; Murakami, T.; Funahashi, K.; Tokunaga, T.; Nihei, K.-i.; Okuno, T.; Kimura, T.; Naoki, H.; Himeno, H. Bioorg. Med. Chem. 2006, 14, 8259–8270. doi:10.1016/j.bmc.2006.09.006
Return to citation in text: [1] -
Corte, J. R.; Fang, T.; Hangeland, J. J.; Friends, T. J.; Rendina, A. R.; Luettgen, J. M.; Bozarth, J. M.; Barbera, F. A.; Rossi, K. A.; Wei, A.; Ramamurthy, V.; Morin, P. E.; Seiffert, D. A.; Wexler, R. R.; Quan, M. L. Bioorg. Med. Chem. Lett. 2015, 25, 925–930. doi:10.1016/j.bmcl.2014.12.050
Return to citation in text: [1] [2] -
Mori, Y.; Ogawa, Y.; Mochikuzi, A.; Nakamura, Y.; Fujimoto, T.; Sugita, C.; Miyazaki, S.; Tamaki, K.; Nagayama, T.; Nagai, Y.; Inoue, S.-i.; Chiba, K.; Nishi, T. Bioorg. Med. Chem. 2013, 21, 5907–5922. doi:10.1016/j.bmc.2013.06.057
Return to citation in text: [1] [2] -
Liu, Y.; Lang, Y.; Patel, N. K.; Ng, G.; Laufer, R.; Li, S.-W.; Edwards, L.; Forrest, B.; Sampson, P. B.; Feher, M.; Ban, F.; Awrey, D. E.; Beletskaya, I.; Mao, G.; Hodgson, R.; Plotnikova, O.; Qiu, W.; Chirgadze, N. Y.; Mason, J. M.; Wei, X.; Lin, D. C.-C.; Che, Y.; Kiarash, R.; Madeira, B.; Fletcher, G. C.; Mak, T. W.; Bray, M. R.; Pauls, H. W. J. Med. Chem. 2015, 58, 3366–3392. doi:10.1021/jm501740a
Return to citation in text: [1] [2] -
Ren, F.; Hogan, P. C.; Anderson, A. J.; Myers, A. G. J. Am. Chem. Soc. 2007, 129, 5381–5383. doi:10.1021/ja071205b
Return to citation in text: [1] -
Shin, C.-g.; Okabe, A.; Ito, A.; Ito, A.; Yonezawa, Y. Bull. Chem. Soc. Jpn. 2002, 75, 1583–1596. doi:10.1246/bcsj.75.1583
Return to citation in text: [1] -
Zou, Y.; Liu, Q.; Deiters, A. Org. Lett. 2011, 13, 4352–4355. doi:10.1021/ol201682k
Return to citation in text: [1] -
Bagley, M. C.; Xiong, X. Org. Lett. 2004, 6, 3401–3404. doi:10.1021/ol0485870
Return to citation in text: [1] -
Jones, M. C.; Marsden, S. P.; Muñoz-Subtil, D. M. Org. Lett. 2006, 8, 5509–5512. doi:10.1021/ol062162r
Return to citation in text: [1] -
Jones, M. C.; Marsden, S. P. Org. Lett. 2008, 10, 4125–4128. doi:10.1021/ol801709c
Return to citation in text: [1] -
Marsden, S. P.; Watson, E. L.; Raw, S. A. Org. Lett. 2008, 10, 2905–2908. doi:10.1021/ol801028e
Return to citation in text: [1] [2] -
Ja, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040–4042. doi:10.1039/b810858g
Return to citation in text: [1] [2] -
Watson, E. L.; Marsden, S. P.; Raw, S. A. Tetrahedron Lett. 2009, 50, 3318–3320. doi:10.1016/j.tetlet.2009.02.076
Return to citation in text: [1] [2] -
Yousif, M. M.; Saeki, S.; Hamana, M. Chem. Pharm. Bull. 1982, 30, 1680–1691. doi:10.1248/cpb.30.1680
Return to citation in text: [1] -
Jones, G.; Pitman, M. A.; Lunt, E.; Lythgoe, D. J.; Abarca, B.; Ballesteros, R.; Elmasnaouy, M. Tetrahedron 1997, 53, 8257–8268. doi:10.1016/S0040-4020(97)00491-2
Return to citation in text: [1] -
Londregan, A. T.; Jennings, S.; Wei, L. Org. Lett. 2011, 13, 1840–1843. doi:10.1021/ol200352g
Return to citation in text: [1] -
Londregan, A. T.; Burford, K.; Conn, E. L.; Hesp, K. D. Org. Lett. 2014, 16, 3336–3339. doi:10.1021/ol501359r
Return to citation in text: [1] -
Lecointre, B.; Azzouz, R.; Bischoff, L. Tetrahedron Lett. 2014, 55, 1913–1915. doi:10.1016/j.tetlet.2014.01.134
Return to citation in text: [1] -
Johnson, T. C.; Marsden, S. P. unpublished observations.
Return to citation in text: [1] -
Edgar, M. T.; Pettit, G. R.; Krupa, T. S. J. Org. Chem. 1979, 44, 396–400. doi:10.1021/jo01317a018
Return to citation in text: [1] -
Herdeis, C.; Gebhard, R. Arch. Pharm. 1987, 320, 546–553. doi:10.1002/ardp.19873200612
Return to citation in text: [1] -
Seebach, D. Angew. Chem., Int. Ed. Engl. 1979, 18, 239–258. doi:10.1002/anie.197902393
Return to citation in text: [1] -
Matsumoto, M.; Harada, M.; Yamashita, Y.; Kobayashi, S. Chem. Commun. 2014, 50, 13041–13044. doi:10.1039/C4CC06156J
Return to citation in text: [1] -
Liu, X.; Gao, A.; Ding, L.; Xu, J.; Zhao, B. Org. Lett. 2014, 16, 2118–2121. doi:10.1021/ol500522d
Return to citation in text: [1] -
Wu, Y.; Hu, L.; Li, Z.; Deng, L. Nature 2015, 523, 445–450. doi:10.1038/nature14617
Return to citation in text: [1]
| 24. | Seebach, D. Angew. Chem., Int. Ed. Engl. 1979, 18, 239–258. doi:10.1002/anie.197902393 |
| 25. | Matsumoto, M.; Harada, M.; Yamashita, Y.; Kobayashi, S. Chem. Commun. 2014, 50, 13041–13044. doi:10.1039/C4CC06156J |
| 26. | Liu, X.; Gao, A.; Ding, L.; Xu, J.; Zhao, B. Org. Lett. 2014, 16, 2118–2121. doi:10.1021/ol500522d |
| 27. | Wu, Y.; Hu, L.; Li, Z.; Deng, L. Nature 2015, 523, 445–450. doi:10.1038/nature14617 |
| 1. | Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337–2347. doi:10.1039/b602413k |
| 5. | Mori, Y.; Ogawa, Y.; Mochikuzi, A.; Nakamura, Y.; Fujimoto, T.; Sugita, C.; Miyazaki, S.; Tamaki, K.; Nagayama, T.; Nagai, Y.; Inoue, S.-i.; Chiba, K.; Nishi, T. Bioorg. Med. Chem. 2013, 21, 5907–5922. doi:10.1016/j.bmc.2013.06.057 |
| 4. | Corte, J. R.; Fang, T.; Hangeland, J. J.; Friends, T. J.; Rendina, A. R.; Luettgen, J. M.; Bozarth, J. M.; Barbera, F. A.; Rossi, K. A.; Wei, A.; Ramamurthy, V.; Morin, P. E.; Seiffert, D. A.; Wexler, R. R.; Quan, M. L. Bioorg. Med. Chem. Lett. 2015, 25, 925–930. doi:10.1016/j.bmcl.2014.12.050 |
| 22. | Edgar, M. T.; Pettit, G. R.; Krupa, T. S. J. Org. Chem. 1979, 44, 396–400. doi:10.1021/jo01317a018 |
| 23. | Herdeis, C.; Gebhard, R. Arch. Pharm. 1987, 320, 546–553. doi:10.1002/ardp.19873200612 |
| 3. | Hashimoto, M.; Murakami, T.; Funahashi, K.; Tokunaga, T.; Nihei, K.-i.; Okuno, T.; Kimura, T.; Naoki, H.; Himeno, H. Bioorg. Med. Chem. 2006, 14, 8259–8270. doi:10.1016/j.bmc.2006.09.006 |
| 13. | Marsden, S. P.; Watson, E. L.; Raw, S. A. Org. Lett. 2008, 10, 2905–2908. doi:10.1021/ol801028e |
| 14. | Ja, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040–4042. doi:10.1039/b810858g |
| 15. | Watson, E. L.; Marsden, S. P.; Raw, S. A. Tetrahedron Lett. 2009, 50, 3318–3320. doi:10.1016/j.tetlet.2009.02.076 |
| 2. | Leet, J. E.; Schroeder, D. R.; Langley, D. R.; Colson, K. L.; Huang, S.; Klohr, S. E.; Lee, M. S.; Golik, J.; Hofstead, S. J.; Doyle, T. W.; Matson, J. A. J. Am. Chem. Soc. 1993, 115, 8432–8443. doi:10.1021/ja00071a062 |
| 16. | Yousif, M. M.; Saeki, S.; Hamana, M. Chem. Pharm. Bull. 1982, 30, 1680–1691. doi:10.1248/cpb.30.1680 |
| 17. | Jones, G.; Pitman, M. A.; Lunt, E.; Lythgoe, D. J.; Abarca, B.; Ballesteros, R.; Elmasnaouy, M. Tetrahedron 1997, 53, 8257–8268. doi:10.1016/S0040-4020(97)00491-2 |
| 18. | Londregan, A. T.; Jennings, S.; Wei, L. Org. Lett. 2011, 13, 1840–1843. doi:10.1021/ol200352g |
| 19. | Londregan, A. T.; Burford, K.; Conn, E. L.; Hesp, K. D. Org. Lett. 2014, 16, 3336–3339. doi:10.1021/ol501359r |
| 20. | Lecointre, B.; Azzouz, R.; Bischoff, L. Tetrahedron Lett. 2014, 55, 1913–1915. doi:10.1016/j.tetlet.2014.01.134 |
| 4. | Corte, J. R.; Fang, T.; Hangeland, J. J.; Friends, T. J.; Rendina, A. R.; Luettgen, J. M.; Bozarth, J. M.; Barbera, F. A.; Rossi, K. A.; Wei, A.; Ramamurthy, V.; Morin, P. E.; Seiffert, D. A.; Wexler, R. R.; Quan, M. L. Bioorg. Med. Chem. Lett. 2015, 25, 925–930. doi:10.1016/j.bmcl.2014.12.050 |
| 7. | Ren, F.; Hogan, P. C.; Anderson, A. J.; Myers, A. G. J. Am. Chem. Soc. 2007, 129, 5381–5383. doi:10.1021/ja071205b |
| 9. | Zou, Y.; Liu, Q.; Deiters, A. Org. Lett. 2011, 13, 4352–4355. doi:10.1021/ol201682k |
| 10. | Bagley, M. C.; Xiong, X. Org. Lett. 2004, 6, 3401–3404. doi:10.1021/ol0485870 |
| 6. | Liu, Y.; Lang, Y.; Patel, N. K.; Ng, G.; Laufer, R.; Li, S.-W.; Edwards, L.; Forrest, B.; Sampson, P. B.; Feher, M.; Ban, F.; Awrey, D. E.; Beletskaya, I.; Mao, G.; Hodgson, R.; Plotnikova, O.; Qiu, W.; Chirgadze, N. Y.; Mason, J. M.; Wei, X.; Lin, D. C.-C.; Che, Y.; Kiarash, R.; Madeira, B.; Fletcher, G. C.; Mak, T. W.; Bray, M. R.; Pauls, H. W. J. Med. Chem. 2015, 58, 3366–3392. doi:10.1021/jm501740a |
| 11. | Jones, M. C.; Marsden, S. P.; Muñoz-Subtil, D. M. Org. Lett. 2006, 8, 5509–5512. doi:10.1021/ol062162r |
| 12. | Jones, M. C.; Marsden, S. P. Org. Lett. 2008, 10, 4125–4128. doi:10.1021/ol801709c |
| 13. | Marsden, S. P.; Watson, E. L.; Raw, S. A. Org. Lett. 2008, 10, 2905–2908. doi:10.1021/ol801028e |
| 14. | Ja, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040–4042. doi:10.1039/b810858g |
| 15. | Watson, E. L.; Marsden, S. P.; Raw, S. A. Tetrahedron Lett. 2009, 50, 3318–3320. doi:10.1016/j.tetlet.2009.02.076 |
| 5. | Mori, Y.; Ogawa, Y.; Mochikuzi, A.; Nakamura, Y.; Fujimoto, T.; Sugita, C.; Miyazaki, S.; Tamaki, K.; Nagayama, T.; Nagai, Y.; Inoue, S.-i.; Chiba, K.; Nishi, T. Bioorg. Med. Chem. 2013, 21, 5907–5922. doi:10.1016/j.bmc.2013.06.057 |
| 6. | Liu, Y.; Lang, Y.; Patel, N. K.; Ng, G.; Laufer, R.; Li, S.-W.; Edwards, L.; Forrest, B.; Sampson, P. B.; Feher, M.; Ban, F.; Awrey, D. E.; Beletskaya, I.; Mao, G.; Hodgson, R.; Plotnikova, O.; Qiu, W.; Chirgadze, N. Y.; Mason, J. M.; Wei, X.; Lin, D. C.-C.; Che, Y.; Kiarash, R.; Madeira, B.; Fletcher, G. C.; Mak, T. W.; Bray, M. R.; Pauls, H. W. J. Med. Chem. 2015, 58, 3366–3392. doi:10.1021/jm501740a |
| 8. | Shin, C.-g.; Okabe, A.; Ito, A.; Ito, A.; Yonezawa, Y. Bull. Chem. Soc. Jpn. 2002, 75, 1583–1596. doi:10.1246/bcsj.75.1583 |
© 2016 Johnson and Marsden; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)