Abstract
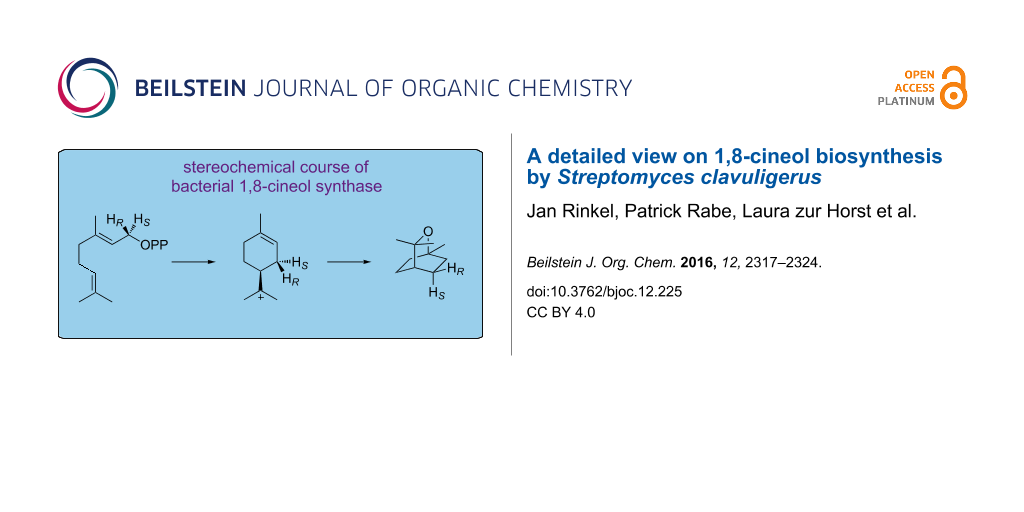
The stereochemical course of the cyclisation reaction catalysed by the bacterial 1,8-cineol synthase from Streptomyces clavuligerus was investigated using stereospecifically deuterated substrates. In contrast to the well investigated plant enzyme from Salvia officinalis, the reaction proceeds via (S)-linalyl diphosphate and the (S)-terpinyl cation, while the final cyclisation reaction is in both cases a syn addition, as could be shown by incubation of (2-13C)geranyl diphosphate in deuterium oxide.

Graphical Abstract
Introduction
Among all classes of natural products the climax of structural diversity and complexity is reached within the largest, the terpenoids. An estimated number of 75,000 different compounds are known from all kinds of organisms including plants [1], bacteria [2-5], fungi [6] and, as recently shown, even social amoebae [7]. These molecules are all made from only a handful of linear and achiral precursors such as geranyl diphosphate (GPP, monoterpenes), farnesyl diphosphate (FPP, sesquiterpenes) and geranylgeranyl diphosphate (GGPP, diterpenes). Terpene cyclases (type I) contain a trinuclear (Mg2+)3 cluster in their active site that is stabilised by binding to several highly conserved motifs including the aspartate-rich motif (DDXXD) and the NSE triad (ND(L,I,V)XSXXXE, modified in plants to a DTE triad: DD(L,I,V)XTXXXE) [8]. Their substrates bind with the diphosphate portion to the (Mg2+)3 cluster and via hydrogen bridges to a highly conserved arginine (diphosphate sensor) and a RY dimer [9]. The substrate is ionised by diphosphate abstraction and the resulting allyl cation undergoes a domino reaction via a series of cationic intermediates and a final deprotonation or attack of water to yield a terpene hydrocarbon or alcohol. This reaction cascade proceeds in a hydrophobic cavity from which water is excluded to enable carbocation chemistry in an aqueous environment. Furthermore, the hydrophobic cavity provides a template that arranges the substrate in a certain conformation to determine the formation of a specific product. Single residues such as phenylalanines are involved in the stabilisation of cationic intermediates, e.g., by cation–π interactions [8-10]. The overall process usually generates an enantiomerically pure (poly)cyclic terpene with several stereogenic centres. A large variety of carbon skeletons is accessible, e g., more than 120 skeletons each representing various stereoisomers and constitutional isomers with different positioning of olefinic double bonds or alcohol functions are known just for sesquiterpenes [11]. The structural diversity of terpenoids can be further increased by the action of tailoring enzymes such as cytochrome P450 monooxygenases and acyl transferases [12,13]. Very few cases are known in which terpene cyclases generate an achiral product as exemplified by the monoterpene 1,8-cineol (eucalyptol, 1) and the sesquiterpenes germacrene B (2) and α-humulene (3) (Figure 1).
![[1860-5397-12-225-1]](/bjoc/content/figures/1860-5397-12-225-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
A direct 1,6-cyclisation of the monoterpene precursor GPP to 1 is prevented by the topological constraints associated with the (2E) geometry which necessitates the isomerisation of GPP (4) to linalyl diphosphate (LPP, 5) followed by an anti,endo-SN’-cyclisation [14], but the stereochemical course of this reaction is not readily clear and may proceed via either enantiomer of the α-terpinyl cation (6, Scheme 1). Isotopic labelling experiments currently experience a revival [15] and are a very powerful method to follow the enzyme mechanisms of terpene cyclases [16-24] including the stereochemical courses of the cyclisation reactions (reviewed in [25]). While the stereochemical course of the GPP cyclisation to 1 has been investigated for the 1,8-cineol synthase from Salvia officinalis [26,27], it is unknown for the bacterial enzyme that was recently reported from Streptomyces clavuligerus [28]. Here we describe isotopic labelling experiments that gave insights into the cyclisation mechanism of the bacterial 1,8-cineol synthase.
![[1860-5397-12-225-i1]](/bjoc/content/inline/1860-5397-12-225-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cyclisation of GPP to 1 via the (R)-terpinyl cation ((R)-6, left) or the (S)-terpinyl cation ((S)-6, right).
Scheme 1: Cyclisation of GPP to 1 via the (R)-terpinyl cation ((R)-6, left) or the (S)-terpinyl cation ((S)-6...
Results
The absolute configuration of the intermediate terpinyl cation
While the two possible cyclisation pathways via (R)- and (S)-6 to 1 cannot be distinguished with unlabelled GPP, its two enantiotopic protons at C-1 (indicated by HR for the pro-R hydrogen and HS for the pro-S hydrogen) end up in diastereotopic positions of 1. Thus, a labelling experiment using the deuterated substrates (R)-(1-2H)GPP (HR = 2H, HS = H) and (S)-(1-2H)GPP (HR = H, HS = 2H) can give insights whether the cyclisation proceeds via (R)- or (S)-6, by determination in which of the distinguishable diastereotopic positions the label ends up. The synthesis of the two enantiomers of (1-2H)GPP (Scheme S1, Supporting Information File 1) was performed by Alpine borane reduction [29] (both enantiomers of this reagent are commercially available) of (1-2H)geranial to (R)- and (S)-(1-2H)geraniol that were obtained with high enantiomeric excess (>95% ee) as determined by Mosher ester analysis (Figure S1, Supporting Information File 1). The alcohols were subsequently converted into the corresponding diphosphates using triethylammonium phosphate in trichloroacetonitrile [30,31]. The gene encoding the 1,8-cineol synthase [28] was cloned into the yeast-to-Escherichia coli shuttle vector pYE-Express by homologous recombination in yeast [32], followed by expression in E. coli BL21. The protein was purified by Ni2+-NTA affinity chromatography and used to convert both (R)- and (S)-(1-2H)GPP into (2H)-1 (in agreement with the findings described in reference [28], 1 is the only product from unlabelled GPP as was shown by GC–MS, Figure S2, Supporting Information File 1). The obtained products were analysed by HSQC spectroscopy (Figure 2). While for unlabelled 1 a 2:2 signal intensity is observed for the crosspeaks representing the two pairs of enantiotopic hydrogens HA and HB connected to carbons C-3 and C-5, the sample obtained from (R)-(1-2H)GPP gave a 1:2 ratio of signal intensities (i.e., HA = 2H), while the sample from (S)-(1-2H)GPP resulted in ratio of 2:1 by peak integration (i.e., HB = 2H), indicating the cyclisation via (S)-LPP ((S)-5) and the (S)-terpinyl cation ((S)-6) (Scheme 1, right).
![[1860-5397-12-225-2]](/bjoc/content/figures/1860-5397-12-225-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Partial HSQC spectra showing the region of crosspeaks for HA and HB connected to C-3 and C-5 of A) unlabelled 1, B) (2H)-1 obtained by enzymatic conversion of (R)-(1-2H)GPP, and C) (2H)-1 obtained by enzymatic conversion of (S)-(1-2H)GPP. The indicated chemical shift data are for unlabelled 1 (for full data cf. Table S1, Supporting Information File 1).
Figure 2: Partial HSQC spectra showing the region of crosspeaks for HA and HB connected to C-3 and C-5 of A) ...
Syn versus anti addition in the final ring closure
The final cyclisation step from (S)-7 via 8 to 1 can in principle proceed either via a syn or an anti addition to the olefinic double bond, requiring a protonation of the original C-2 of GPP. To distinguish between these alternatives (2-13C)GPP was synthesised from sulcatone (Scheme S2, Supporting Information File 1) and converted by the 1,8-cineol synthase in deuterium oxide. The obtained product was analysed by HSQC spectroscopy (Figure 3A), showing that deuterium is taken up into the exo position at the 13C-labelled C-2 of 1 (indicated by H’), while the endo position (H’’) is occupied by the proton from the substrate, resulting in a strong crosspeak [22]. Furthermore, deuterium incorporation at C-2 was indicated by a strongly enhanced triplet in the 13C NMR spectrum due to 13C-2H-spin coupling (Figure S4, Supporting Information File 1) [11,20,23]. This finding is in agreement with a syn addition to the olefinic double bond of (S)-7 in the final cyclisation step. It is possible that the proton is directly transfered from the protonated hydroxy function in (S)-7 to C-2 (Figure 3B). Alternatively, a deprotonation of (S)-7 to the hypothetical neutral intermediate α-terpineol followed by reprotonation at C-2 from the Si face can be assumed, but these two alternatives cannot be distinguished based on the labelling experiments described here.
![[1860-5397-12-225-3]](/bjoc/content/figures/1860-5397-12-225-3.png?scale=1.7&max-width=1024&background=FFFFFF)
Figure 3: A) Partial HSQC spectrum showing the region of crosspeaks of C-2 with its directly connected hydrogens H’ and H’’ for compound (2-13C,2-2H)-1 obtained by enzymatic conversion of (2-13C)GPP in deuterium oxide. Deuterium was specifically incorporated into H’ position. The indicated chemical shift data are for unlabelled 1 (for full data cf. Table S1, Supporting Information File 1). Blue circles point to 13C-labelled carbons. B) Intramolecular proton transfer from the protonated hydroxy function in (S)-7 to C-2.
Figure 3: A) Partial HSQC spectrum showing the region of crosspeaks of C-2 with its directly connected hydrog...
Discussion
Plant and bacterial terpene cyclases show important structural differences [33]. While plant monoterpene synthases are composed of α and β domains and exhibit a quarternary α2β2 structure [34,35], bacterial mono- and sesquiterpene cyclases are monodomain enzymes (α) [9,10,36]. Accordingly, also the 1,8-cineol synthases from Salvia officinalis and from Streptomyces clavuligerus are not related and have evolved independently. While the plant enzyme was shown to convert GPP via (R)-LPP ((R)-5) and the (R)-terpinyl cation ((R)-6) into 1 [27], the experiments described here revealed a different course for the bacterial enzyme via (S)-6. This finding is particularly interesting, because it reflects the frequent observation that the (chiral) products of bacterial terpene cyclases represent the opposite enantiomers as usually generated by plant enzymes [24,37,38]. Both enzymes from Salvia officinalis and from Streptomyces clavuligerus share the syn addition in the final cyclisation step which can be rationalised by a direct intramolecular proton transfer, circumventing the need of a low-energy neutral intermediate such as α-terpineol. However, in case of the sesquiterpene ethers corvol ethers A (19) and B (18) a reprotonation step was shown to proceed from the opposite face than the preceeding attack of water, thus excluding a direct proton transfer from oxygen to the neighbouring carbon (reactions from 12 to 14 in Scheme 2). Conclusively, reprotonation of the neutral intermediate 13 is possible in this case [21].
![[1860-5397-12-225-i2]](/bjoc/content/inline/1860-5397-12-225-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanism for the cyclisation of FPP to corvol ethers A (19) and B (18). WMR: Wagner-Meerwein rearrangement.
Scheme 2: Mechanism for the cyclisation of FPP to corvol ethers A (19) and B (18). WMR: Wagner-Meerwein rearr...
Experimental
Cloning and homologous recombination
Cells of Streptomyces clavuligerus ATCC 27064 were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Cultivation was done in liquid 65 Gym medium (4 g yeast extract, 4 g glucose, 10 g malt extract, 1 L water, pH 7.2) and isolation of genomic DNA was performed using a standard protocol. The gene WP_003952918 encoding the 1,8-cineol synthase was amplified using forward primer (ATGCCCGCCGGCCACGAAGA) and reversed primer (TCACCAAGGGGTGGTGGCCC). The isolated PCR product was elongated for homologous recombination with pYE-Express in yeast with a second set of primers (GGCAGCCATATGGCTAGCATGACTGGTGGAATGCCCGCCGGCCACGAAGA and TCTCAGTGGTGGTGGTGGTGGTGCTCGAGTTCACCAAGGGGTGGTGG CCC). This elongated product was transformed in Saccharomyces cerevisiae FY834 together with linearised vector pYE-Express [32] (EcoRI and HindIII digestion) using the LiOAc/SS carrier DNA protocol [39]. Transformed cells were plated on SM-URA medium (20 g glucose, 1.7 g yeast nitrogen base, 5 g ammonium sulphate, 0.77 g nutritional supplement minus uracil, 24 g agar, 1 L water) and grown for 3 days at 28 °C. Plasmids were isolated using the kit Zymoprep Yeast Plasmid Miniprep II (Zymo Research, Irvine, USA), shuttled in E. coli BL21 by electroporation and confirmed by sequencing.
Incubation experiments with (1R)- and (1S)-(1-2H)GPP
A preculture of E. coli BL21 cells carrying the plasmid pYE_WP003952918 in 2YT medium (16 g trypton, 10 g yeast extract, 5 g NaCl, 1 L water, pH 7.2) was grown overnight to inoculate a 2YT main culture (2 L). The cultures were shaken at 160 rpm, 37 °C until OD600 = 0.4 was reached. Prior to induction with IPTG (0.4 mM), the cultures were cooled down to 18 °C. After incubation overnight (160 rpm, 18 °C), cells were harvested by centrifugation (5000g, 4 °C, 45 min) and resuspended in binding buffer (20 mL; 20 mM Na2HPO4, 0.5 mM NaCl, 20 mM imidazole, 1 mM MgCl2, pH 7.0). The cells were crushed by ultra-sonification (6 × 1 min, 4 °C) and the cell debris pellet was separated by centrifugation (10 min, 15500g, 4 °C). The soluble fraction was loaded onto a Ni2+-NTA affinity column (Novagen) and treated with binding buffer (2 × 10 mL). The target protein was then eluted with elution buffer (2 × 10 mL; 20 mM Na2HPO4, 0.5 M NaCl, 0.5 M imidazole, 1 mM MgCl2, pH 7.0) and used directly for incubations with 5 mg of (1R)- and (1S)-(1-2H)GPP, solved in incubation buffer (50 mM Tris/HCl, 10 mM MgCl2, 20% v/v glycerol, pH 8.2) to reach a final substrate concentration of 0.2 mg/mL. The enzyme reaction was incubated for 2 h at 28 h, overlaid with 400 μL (2H6)benzene and further incubated overnight. The organic phase was separated, dried over MgSO4 and directly analysed by GC–MS and NMR.
Incubation experiment with (2-13C)GPP
Enzyme purification starting from an E. coli expression culture (0.5 L) was performed as described above. The last washing fraction was substituted with 2H2O-based binding buffer and elution was done with 2H2O-based elution buffer. The first elution fraction was incubated with (2-13C)GPP (0.8 mg) for 16 h at 28 °C. The enzyme reaction was extracted with (2H6)benzene (0.6 mL), dried with MgSO4 and the extract was analysed directly by GC–MS and NMR.
NMR spectroscopy
To record NMR spectra, instruments AV Avance DMX-500 (500 MHz), DPX-400 (400 MHz) and AV III HD Cryo (700 MHz) from Bruker were used. Solvent signals were used to reference the spectra (1H NMR, residual proton signals: (2H6)benzene δ = 7.16; 13C NMR: (2H6)benzene δ = 128.06) [40].
GC–MS analysis
An Agilent 7890B gas chromatograph equipped with a HP5-MS silica column (30 m, 0.25 mm inner diameter, 0.50 μm film) connected to an Agilent 5977A inert mass selective detector was used to acquire GC–MS data. Instrumental settings were: (1) inlet pressure: 77.1 kPa, He: 23.3 mL/min, (2) transfer line: 250 °C, (3) electron energy: 70 eV. The GC was set to 50 °C starting temperature for 5 min, then increasing with 5 °C per minute to 320 °C and holding this temperature for another 5 min. The injection volume was 2 μL and the inlet was operating in split mode (10:1, 60 s valve time). Helium was used as the carrier gas at 1 mL/min. Retention indices were determined against a homologous series of n-alkanes (C8–C40).
Synthesis of (2-13C)geranyl diphosphate
(2-13C)Geraniol was synthesised as reported previously [41]. The synthetic (2-13C)geraniol (16 mg, 0.072 mmol, 1.0 equiv) was dissolved in dry THF (0.3 mL) and PBr3 (8.1 mg, 0.029 mmol, 0.4 equiv) was added at 0 °C. The solution was stirred for 45 min at room temperature. The reaction mixture was hydrolyzed by addition of ice cold water and extracted three times with pentane. The combined organic layers were dried over MgSO4 and the solvent was removed under reduced pressure. The crude product was used for phosphorylation.
In a second flask, to a solution of (n-Bu4)3HP2O7 (97 mg, 0.11 mmol, 1.5 equiv) in dry CH3CN (1.0 mL) the crude product of the allyl bromide (1.0 equiv) was added and the reaction mixture was stirred for 2 h at room temperature and then concentrated under reduced pressure. The colorless oil was loaded onto an ion exchange column (DOWEX 50W-X8, NH4+ form). Elution of the product was performed by addition of two column volumes of ion exchange buffer (0.03 M NH4HCO3 in 2% iPrOH/H2O). Freeze drying yielded the product as a white solid (14.1 mg, 0.04 mmol, 55%).
1H NMR (500 MHz, H2O) δ 5.37 (dt, 1J(C,H) = 156.7 Hz, 3J(H,H) = 6.9 Hz, 1H, CH), 5.16–5.11 (m, 1H, 1 × CH), 4.44–4.38 (m, 2H, 1 × CH2), 2.12–2.06 (m, 2H, 1 × CH2), 2.06–2.00 (m, 2H, 1 × CH2), 1.65 (d, 3J(C,H) = 5.2 Hz, 3H, 1 × CH3), 1.62 (s, 3H, 1 × CH3), 1.56 (s, 3H, 1 × CH3) ppm; 13C NMR (125 MHz, H2O) δ 141.9 (d, 1J(C,C) = 72.7 Hz, 1 × Cq), 133.7 (1 × Cq), 124.1 (1 × CH), 119.6 (d, 3J(P,C) = 8.2 Hz, 1 × 13CH), 38.8 (d, 2J(C,C) = 2.7 Hz, 1 × CH2), 25.6 (d, 3J(C,C) = 2.9 Hz, 1 × CH2), 24.8 (1 × CH3), 16.9 (1 × CH3), 15.6 (d, 2J(C,C) = 1.2 Hz, 1 × CH3) ppm; 31P NMR (202 MHz, H2O) δ −10.0 (m, 1 × P), −10.6 (m, 1 × P) ppm.
Supporting Information
Synthesis schemes, Mosher ester analysis of (R)- and (S)-(1-2H)GPP, gas chromatogram of the enzyme product of 1,8-cineol synthase, 13C NMR of the enzyme product from (2-13C)GPP in deuterium oxide buffer, and full NMR data of 1.
| Supporting Information File 1: Additional material. | ||
| Format: PDF | Size: 446.5 KB | Download |
References
-
Degenhardt, J.; Köllner, T. G.; Gershenzon, J. Phytochemistry 2009, 70, 1621–1637. doi:10.1016/j.phytochem.2009.07.030
Return to citation in text: [1] -
Citron, C. A.; Gleitzmann, J.; Laurenzano, G.; Pukall, R.; Dickschat, J. S. ChemBioChem 2012, 13, 202–214. doi:10.1002/cbic.201100641
Return to citation in text: [1] -
Rabe, P.; Citron, C. A.; Dickschat, J. S. ChemBioChem 2013, 14, 2345–2354. doi:10.1002/cbic.201300329
Return to citation in text: [1] -
Citron, C. A.; Barra, L.; Wink, J.; Dickschat, J. S. Org. Biomol. Chem. 2015, 13, 2673–2683. doi:10.1039/C4OB02609H
Return to citation in text: [1] -
Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-ya, K.; Omura, S.; Cane, D. E.; Ikeda, H. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 857–862. doi:10.1073/pnas.1422108112
Return to citation in text: [1] -
Quin, M. B.; Flynn, C. M.; Schmidt-Dannert, C. Nat. Prod. Rep. 2014, 31, 1449–1473. doi:10.1039/C4NP00075G
Return to citation in text: [1] -
Chen, X.; Köllner, T. G.; Jia, Q.; Norris, A.; Santhanam, B.; Rabe, P.; Dickschat, J. S.; Shaulsky, G.; Gershenzon, J.; Chen, F. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12132–12137. doi:10.1073/pnas.1610379113
Return to citation in text: [1] -
Starks, C. M.; Back, K.; Chappell, J.; Noel, J. P. Science 1997, 277, 1815–1820. doi:10.1126/science.277.5333.1815
Return to citation in text: [1] [2] -
Baer, P.; Rabe, P.; Fischer, K.; Citron, C. A.; Klapschinski, T. A.; Groll, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2014, 53, 7652–7656. doi:10.1002/anie.201403648
Return to citation in text: [1] [2] [3] -
Lesburg, C. A.; Zhai, G.; Cane, D. E.; Christianson, D. W. Science 1997, 277, 1820–1824. doi:10.1126/science.277.5333.1820
Return to citation in text: [1] [2] -
Klapschinski, T. A.; Rabe, P.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 10141–10144. doi:10.1002/anie.201605425
Return to citation in text: [1] [2] -
Bogazkaya, A. M.; von Bühler, C. J.; Kriening, S.; Busch, A.; Seifert, A.; Pleiss, J.; Laschat, S.; Urlacher, V. B. Beilstein J. Org. Chem. 2014, 10, 1347–1353. doi:10.3762/bjoc.10.137
Return to citation in text: [1] -
Huber, T.; Weisheit, L.; Magauer, T. Beilstein J. Org. Chem. 2015, 11, 2521–2539. doi:10.3762/bjoc.11.273
Return to citation in text: [1] -
Croteau, R.; Alonso, W. R.; Koepp, A. E.; Johnson, M. A. Arch. Biochem. Biophys. 1994, 309, 184–192. doi:10.1006/abbi.1994.1101
Return to citation in text: [1] -
Rinkel, J.; Dickschat, J. S. Beilstein J. Org. Chem. 2015, 11, 2493–2508. doi:10.3762/bjoc.11.271
Return to citation in text: [1] -
Meguro, A.; Motoyoshi, Y.; Teramoto, K.; Ueda, S.; Totsuka, Y.; Ando, Y.; Tomita, T.; Kim, S.-Y.; Kimura, T.; Igarashi, M.; Sawa, R.; Shinada, T.; Nishiyama, M.; Kuzuyama, T. Angew. Chem., Int. Ed. 2015, 54, 4353–4356. doi:10.1002/anie.201411923
Return to citation in text: [1] -
Matsuda, Y.; Mitsuhashi, T.; Lee, S.; Hoshino, N.; Mori, T.; Okada, M.; Zhang, H.; Hayashi, F.; Fujita, M.; Abe, I. Angew. Chem., Int. Ed. 2016, 55, 5785–5788. doi:10.1002/anie.201601448
Return to citation in text: [1] -
Okada, M.; Matsuda, Y.; Mitsuhashi, T.; Hoshino, S.; Mori, T.; Nakagawa, K.; Quan, Z.; Qin, B.; Zhang, H.; Hayashi, F.; Kawaide, H.; Abe, I. J. Am. Chem. Soc. 2016, 138, 10011–10018. doi:10.1021/jacs.6b05799
Return to citation in text: [1] -
Ye, Y.; Minami, A.; Mandi, A.; Liu, C.; Taniguchi, T.; Kuzuyama, T.; Monde, K.; Gomi, K.; Oikawa, H. J. Am. Chem. Soc. 2015, 137, 11846–11853. doi:10.1021/jacs.5b08319
Return to citation in text: [1] -
Burkhardt, I.; Siemon, T.; Henrot, M.; Studt, L.; Rösler, S.; Tudzynski, B.; Christmann, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 8748–8751. doi:10.1002/anie.201603782
Return to citation in text: [1] [2] -
Rabe, P.; Janusko, A.; Goldfuss, B.; Dickschat, J. S. ChemBioChem 2016, 17, 146–149. doi:10.1002/cbic.201500543
Return to citation in text: [1] [2] -
Rabe, P.; Rinkel, J.; Klapschinski, T. A.; Barra, L.; Dickschat, J. S. Org. Biomol. Chem. 2016, 14, 158–164. doi:10.1039/C5OB01998B
Return to citation in text: [1] [2] -
Rabe, P.; Pahirulzaman, K. A. K.; Dickschat, J. S. Angew. Chem., Int. Ed. 2015, 54, 6041–6045. doi:10.1002/anie.201501119
Return to citation in text: [1] [2] -
Rabe, P.; Schmitz, T.; Dickschat, J. S. Beilstein J. Org. Chem. 2016, 12, 1839–1850. doi:10.3762/bjoc.12.173
Return to citation in text: [1] [2] -
Dickschat, J. S. Nat. Prod. Rep. 2011, 28, 1917–1936. doi:10.1039/c1np00063b
Return to citation in text: [1] -
Wise, M. L.; Savage, T. J.; Katahira, E.; Croteau, R. J. Biol. Chem. 1998, 273, 14891–14899. doi:10.1074/jbc.273.24.14891
Return to citation in text: [1] -
Wise, M. L.; Urbansky, M.; Helms, G. L.; Coates, R. M.; Croteau, R. J. Am. Chem. Soc. 2002, 124, 8546–8547. doi:10.1021/ja0265714
Return to citation in text: [1] [2] -
Nakano, C.; Kim, H.-K.; Ohnishi, Y. ChemBioChem 2011, 12, 1988–1991. doi:10.1002/cbic.201100330
Return to citation in text: [1] [2] [3] -
Edelstein, R. L.; Weller, V. A.; Distefano, M. D.; Tung, J. S. J. Org. Chem. 1998, 63, 5298–5299. doi:10.1021/jo980304s
Return to citation in text: [1] -
Thulasiram, H. V.; Phan, R. M.; Rivera, S. B.; Poulter, C. D. J. Org. Chem. 2006, 71, 1739–1741. doi:10.1021/jo052384n
Return to citation in text: [1] -
Keller, R. K.; Thompson, R. J. Chromatogr. A 1993, 645, 161–167. doi:10.1016/0021-9673(93)80630-Q
Return to citation in text: [1] -
Dickschat, J. S.; Pahirulzaman, K. A. K.; Rabe, P.; Klapschinski, T. A. ChemBioChem 2014, 15, 810–814. doi:10.1002/cbic.201300763
Return to citation in text: [1] [2] -
Oldfield, E.; Lin, F.-Y. Angew. Chem., Int. Ed. 2012, 51, 1124–1137. doi:10.1002/anie.201103110
Return to citation in text: [1] -
Whittington, D. A.; Wise, M. L.; Urbansky, M.; Coates, R. M.; Croteau, R. B.; Christianson, D. W. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15375–15380. doi:10.1073/pnas.232591099
Return to citation in text: [1] -
Hyatt, D. C.; Youn, B.; Zhao, Y.; Santhamma, B.; Coates, R. M.; Croteau, R. B.; Kang, C. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5360–5365. doi:10.1073/pnas.0700915104
Return to citation in text: [1] -
Baer, P.; Rabe, P.; Citron, C. A.; de Oliveira Mann, C. C.; Kaufmann, N.; Groll, M.; Dickschat, J. S. ChemBioChem 2014, 15, 213–216. doi:10.1002/cbic.201300708
Return to citation in text: [1] -
Ding, L.; Goerls, H.; Dornblut, K.; Lin, W.; Maier, A.; Fiebig, H.-H.; Hertweck, C. J. Nat. Prod. 2015, 78, 2963–2967. doi:10.1021/acs.jnatprod.5b00674
Return to citation in text: [1] -
Dickschat, J. S. Nat. Prod. Rep. 2016, 33, 87–110. doi:10.1039/C5NP00102A
Return to citation in text: [1] -
Gietz, R. D.; Schiestl, R. H. Nat. Protoc. 2007, 2, 31–34. doi:10.1038/nprot.2007.13
Return to citation in text: [1] -
Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. doi:10.1021/om100106e
Return to citation in text: [1] -
Rabe, P.; Barra, L.; Rinkel, J.; Riclea, R.; Citron, C. A.; Klapschinski, T. A.; Janusko, A.; Dickschat, J. S. Angew. Chem., Int. Ed. 2015, 54, 13448–13451. doi:10.1002/anie.201507615
Return to citation in text: [1]
| 21. | Rabe, P.; Janusko, A.; Goldfuss, B.; Dickschat, J. S. ChemBioChem 2016, 17, 146–149. doi:10.1002/cbic.201500543 |
| 32. | Dickschat, J. S.; Pahirulzaman, K. A. K.; Rabe, P.; Klapschinski, T. A. ChemBioChem 2014, 15, 810–814. doi:10.1002/cbic.201300763 |
| 39. | Gietz, R. D.; Schiestl, R. H. Nat. Protoc. 2007, 2, 31–34. doi:10.1038/nprot.2007.13 |
| 1. | Degenhardt, J.; Köllner, T. G.; Gershenzon, J. Phytochemistry 2009, 70, 1621–1637. doi:10.1016/j.phytochem.2009.07.030 |
| 8. | Starks, C. M.; Back, K.; Chappell, J.; Noel, J. P. Science 1997, 277, 1815–1820. doi:10.1126/science.277.5333.1815 |
| 28. | Nakano, C.; Kim, H.-K.; Ohnishi, Y. ChemBioChem 2011, 12, 1988–1991. doi:10.1002/cbic.201100330 |
| 7. | Chen, X.; Köllner, T. G.; Jia, Q.; Norris, A.; Santhanam, B.; Rabe, P.; Dickschat, J. S.; Shaulsky, G.; Gershenzon, J.; Chen, F. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12132–12137. doi:10.1073/pnas.1610379113 |
| 29. | Edelstein, R. L.; Weller, V. A.; Distefano, M. D.; Tung, J. S. J. Org. Chem. 1998, 63, 5298–5299. doi:10.1021/jo980304s |
| 6. | Quin, M. B.; Flynn, C. M.; Schmidt-Dannert, C. Nat. Prod. Rep. 2014, 31, 1449–1473. doi:10.1039/C4NP00075G |
| 2. | Citron, C. A.; Gleitzmann, J.; Laurenzano, G.; Pukall, R.; Dickschat, J. S. ChemBioChem 2012, 13, 202–214. doi:10.1002/cbic.201100641 |
| 3. | Rabe, P.; Citron, C. A.; Dickschat, J. S. ChemBioChem 2013, 14, 2345–2354. doi:10.1002/cbic.201300329 |
| 4. | Citron, C. A.; Barra, L.; Wink, J.; Dickschat, J. S. Org. Biomol. Chem. 2015, 13, 2673–2683. doi:10.1039/C4OB02609H |
| 5. | Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-ya, K.; Omura, S.; Cane, D. E.; Ikeda, H. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 857–862. doi:10.1073/pnas.1422108112 |
| 26. | Wise, M. L.; Savage, T. J.; Katahira, E.; Croteau, R. J. Biol. Chem. 1998, 273, 14891–14899. doi:10.1074/jbc.273.24.14891 |
| 27. | Wise, M. L.; Urbansky, M.; Helms, G. L.; Coates, R. M.; Croteau, R. J. Am. Chem. Soc. 2002, 124, 8546–8547. doi:10.1021/ja0265714 |
| 12. | Bogazkaya, A. M.; von Bühler, C. J.; Kriening, S.; Busch, A.; Seifert, A.; Pleiss, J.; Laschat, S.; Urlacher, V. B. Beilstein J. Org. Chem. 2014, 10, 1347–1353. doi:10.3762/bjoc.10.137 |
| 13. | Huber, T.; Weisheit, L.; Magauer, T. Beilstein J. Org. Chem. 2015, 11, 2521–2539. doi:10.3762/bjoc.11.273 |
| 15. | Rinkel, J.; Dickschat, J. S. Beilstein J. Org. Chem. 2015, 11, 2493–2508. doi:10.3762/bjoc.11.271 |
| 11. | Klapschinski, T. A.; Rabe, P.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 10141–10144. doi:10.1002/anie.201605425 |
| 16. | Meguro, A.; Motoyoshi, Y.; Teramoto, K.; Ueda, S.; Totsuka, Y.; Ando, Y.; Tomita, T.; Kim, S.-Y.; Kimura, T.; Igarashi, M.; Sawa, R.; Shinada, T.; Nishiyama, M.; Kuzuyama, T. Angew. Chem., Int. Ed. 2015, 54, 4353–4356. doi:10.1002/anie.201411923 |
| 17. | Matsuda, Y.; Mitsuhashi, T.; Lee, S.; Hoshino, N.; Mori, T.; Okada, M.; Zhang, H.; Hayashi, F.; Fujita, M.; Abe, I. Angew. Chem., Int. Ed. 2016, 55, 5785–5788. doi:10.1002/anie.201601448 |
| 18. | Okada, M.; Matsuda, Y.; Mitsuhashi, T.; Hoshino, S.; Mori, T.; Nakagawa, K.; Quan, Z.; Qin, B.; Zhang, H.; Hayashi, F.; Kawaide, H.; Abe, I. J. Am. Chem. Soc. 2016, 138, 10011–10018. doi:10.1021/jacs.6b05799 |
| 19. | Ye, Y.; Minami, A.; Mandi, A.; Liu, C.; Taniguchi, T.; Kuzuyama, T.; Monde, K.; Gomi, K.; Oikawa, H. J. Am. Chem. Soc. 2015, 137, 11846–11853. doi:10.1021/jacs.5b08319 |
| 20. | Burkhardt, I.; Siemon, T.; Henrot, M.; Studt, L.; Rösler, S.; Tudzynski, B.; Christmann, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 8748–8751. doi:10.1002/anie.201603782 |
| 21. | Rabe, P.; Janusko, A.; Goldfuss, B.; Dickschat, J. S. ChemBioChem 2016, 17, 146–149. doi:10.1002/cbic.201500543 |
| 22. | Rabe, P.; Rinkel, J.; Klapschinski, T. A.; Barra, L.; Dickschat, J. S. Org. Biomol. Chem. 2016, 14, 158–164. doi:10.1039/C5OB01998B |
| 23. | Rabe, P.; Pahirulzaman, K. A. K.; Dickschat, J. S. Angew. Chem., Int. Ed. 2015, 54, 6041–6045. doi:10.1002/anie.201501119 |
| 24. | Rabe, P.; Schmitz, T.; Dickschat, J. S. Beilstein J. Org. Chem. 2016, 12, 1839–1850. doi:10.3762/bjoc.12.173 |
| 8. | Starks, C. M.; Back, K.; Chappell, J.; Noel, J. P. Science 1997, 277, 1815–1820. doi:10.1126/science.277.5333.1815 |
| 9. | Baer, P.; Rabe, P.; Fischer, K.; Citron, C. A.; Klapschinski, T. A.; Groll, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2014, 53, 7652–7656. doi:10.1002/anie.201403648 |
| 10. | Lesburg, C. A.; Zhai, G.; Cane, D. E.; Christianson, D. W. Science 1997, 277, 1820–1824. doi:10.1126/science.277.5333.1820 |
| 40. | Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. Organometallics 2010, 29, 2176–2179. doi:10.1021/om100106e |
| 9. | Baer, P.; Rabe, P.; Fischer, K.; Citron, C. A.; Klapschinski, T. A.; Groll, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2014, 53, 7652–7656. doi:10.1002/anie.201403648 |
| 14. | Croteau, R.; Alonso, W. R.; Koepp, A. E.; Johnson, M. A. Arch. Biochem. Biophys. 1994, 309, 184–192. doi:10.1006/abbi.1994.1101 |
| 41. | Rabe, P.; Barra, L.; Rinkel, J.; Riclea, R.; Citron, C. A.; Klapschinski, T. A.; Janusko, A.; Dickschat, J. S. Angew. Chem., Int. Ed. 2015, 54, 13448–13451. doi:10.1002/anie.201507615 |
| 32. | Dickschat, J. S.; Pahirulzaman, K. A. K.; Rabe, P.; Klapschinski, T. A. ChemBioChem 2014, 15, 810–814. doi:10.1002/cbic.201300763 |
| 30. | Thulasiram, H. V.; Phan, R. M.; Rivera, S. B.; Poulter, C. D. J. Org. Chem. 2006, 71, 1739–1741. doi:10.1021/jo052384n |
| 31. | Keller, R. K.; Thompson, R. J. Chromatogr. A 1993, 645, 161–167. doi:10.1016/0021-9673(93)80630-Q |
| 28. | Nakano, C.; Kim, H.-K.; Ohnishi, Y. ChemBioChem 2011, 12, 1988–1991. doi:10.1002/cbic.201100330 |
| 27. | Wise, M. L.; Urbansky, M.; Helms, G. L.; Coates, R. M.; Croteau, R. J. Am. Chem. Soc. 2002, 124, 8546–8547. doi:10.1021/ja0265714 |
| 24. | Rabe, P.; Schmitz, T.; Dickschat, J. S. Beilstein J. Org. Chem. 2016, 12, 1839–1850. doi:10.3762/bjoc.12.173 |
| 37. | Ding, L.; Goerls, H.; Dornblut, K.; Lin, W.; Maier, A.; Fiebig, H.-H.; Hertweck, C. J. Nat. Prod. 2015, 78, 2963–2967. doi:10.1021/acs.jnatprod.5b00674 |
| 38. | Dickschat, J. S. Nat. Prod. Rep. 2016, 33, 87–110. doi:10.1039/C5NP00102A |
| 34. | Whittington, D. A.; Wise, M. L.; Urbansky, M.; Coates, R. M.; Croteau, R. B.; Christianson, D. W. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15375–15380. doi:10.1073/pnas.232591099 |
| 35. | Hyatt, D. C.; Youn, B.; Zhao, Y.; Santhamma, B.; Coates, R. M.; Croteau, R. B.; Kang, C. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5360–5365. doi:10.1073/pnas.0700915104 |
| 9. | Baer, P.; Rabe, P.; Fischer, K.; Citron, C. A.; Klapschinski, T. A.; Groll, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2014, 53, 7652–7656. doi:10.1002/anie.201403648 |
| 10. | Lesburg, C. A.; Zhai, G.; Cane, D. E.; Christianson, D. W. Science 1997, 277, 1820–1824. doi:10.1126/science.277.5333.1820 |
| 36. | Baer, P.; Rabe, P.; Citron, C. A.; de Oliveira Mann, C. C.; Kaufmann, N.; Groll, M.; Dickschat, J. S. ChemBioChem 2014, 15, 213–216. doi:10.1002/cbic.201300708 |
| 11. | Klapschinski, T. A.; Rabe, P.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 10141–10144. doi:10.1002/anie.201605425 |
| 20. | Burkhardt, I.; Siemon, T.; Henrot, M.; Studt, L.; Rösler, S.; Tudzynski, B.; Christmann, M.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 8748–8751. doi:10.1002/anie.201603782 |
| 23. | Rabe, P.; Pahirulzaman, K. A. K.; Dickschat, J. S. Angew. Chem., Int. Ed. 2015, 54, 6041–6045. doi:10.1002/anie.201501119 |
| 33. | Oldfield, E.; Lin, F.-Y. Angew. Chem., Int. Ed. 2012, 51, 1124–1137. doi:10.1002/anie.201103110 |
| 28. | Nakano, C.; Kim, H.-K.; Ohnishi, Y. ChemBioChem 2011, 12, 1988–1991. doi:10.1002/cbic.201100330 |
| 22. | Rabe, P.; Rinkel, J.; Klapschinski, T. A.; Barra, L.; Dickschat, J. S. Org. Biomol. Chem. 2016, 14, 158–164. doi:10.1039/C5OB01998B |
© 2016 Rinkel et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)