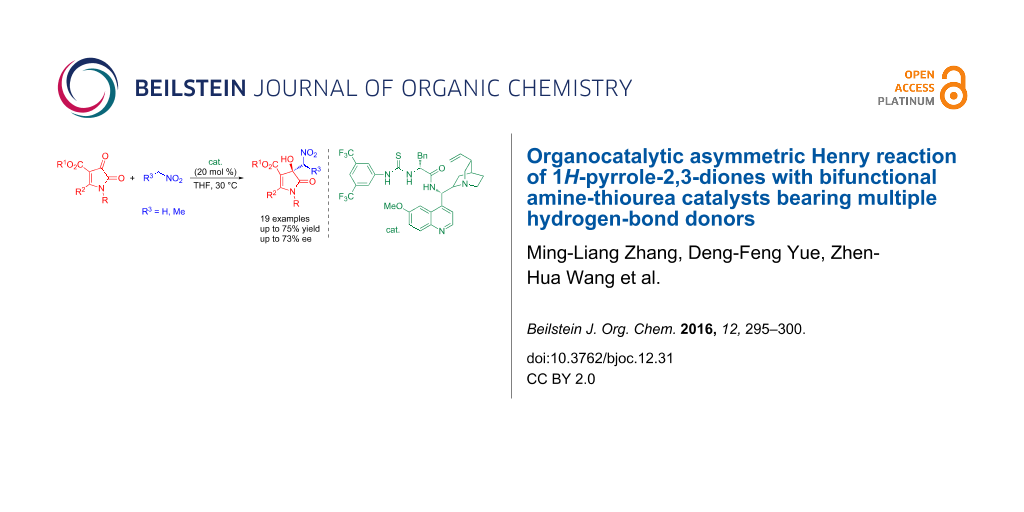
Abstract
For the first time, a catalytic asymmetric Henry reaction of 1H-pyrrole-2,3-diones was achieved with a chiral bifunctional amine-thiourea as a catalyst possessing multiple hydrogen-bond donors. With this developed method, a range of 3-hydroxy-3-nitromethyl-1H-pyrrol-2(3H)-ones bearing quaternary stereocenters were obtained in acceptable yield (up to 75%) and enantioselectivity (up to 73% ee).

Graphical Abstract
Introduction
Asymmetric organocatalysis has been demonstrated to be an effective and versatile strategy in facilitating a variety of organic transformations over the past decade, and numerous catalytic asymmetric reactions have been developed with various activation modes [1-6]. In this realm, chiral bifunctional catalysts, possessing two active sites, have captured tremendous attention in particular due to their unique ability of the simultaneous activation of the nucleophile and the electrophile in the same transition state [7-11]. Among them, chiral bifunctional thioureas bearing multiple hydrogen-bond donors have been successfully used as chiral organocatalysts for the asymmetric Michael addition and Mannich reactions [12-14]. Meanwhile, the Henry reaction is one of the most important carbon–carbon bond-forming reactions that provides straightforward access to β-nitroalcohols, which can be further transformed into amino-alcohols, amino acids and carbonyl compounds [15]. Much attention has been devoted to the development of an efficient catalytic asymmetric version of this reaction from readily accessible nitroalkanes and carbonyl compounds [16], such as aldehydes [17-19], α-ketoesters [20], α-ketophosphonates [21,22], fluoromethyl ketones [23,24] and isatins [25,26]. Despite these significant advances described above, the use of more challenging ketones with heterocyclic structures as Henry acceptors has remained relatively less explored. In this context, developing a new Henry reaction for the construction of the useful and versatile β-nitroalcohol scaffolds is still desirable.
Pyrrole skeletons represent an important class of heterocycles and are frequently found in many biologically active molecules and natural products [27,28]. Particularly, 3-substituted-3-hydroxy-1H-pyrrol-2(3H)-one derivatives exhibit a wide spectrum of biological activities [29]. The reaction of 1H-pyrrole-2,3-diones with various nucleophiles should be a straightforward way to access diverse and interesting 3-substituted-3-hydroxy-1H-pyrrol-2(3H)-ones. A survey of the literature reveals that the study of 1H-pyrrole-2,3-diones is mainly focused on three-component spiro-heterocyclization reactions [30-32]. However, for the catalytic asymmetric transformation, only one example of an aldol reaction of 1H-pyrrole-2,3-diones with ketones has been reported so far (Scheme 1) [33]. Recently, our group developed a chiral bifunctional multiple hydrogen-bond amine-thiourea-catalyzed Michael reaction of acetyl phosphonates with nitroolefins, giving a series of β-substituted nitro compounds with excellent stereoselectivity [34]. Therefore, as part of our research program aimed at establishing new methods for the construction of quaternary stereocenters [35-37], we envisioned that the Henry reaction of nitroalkanes with 1H-pyrrole-2,3-diones should take place with a chiral bifunctional amine-thiourea catalyst, leading to 3-hydroxy-3-nitromethyl-1H-pyrrol-2(3H)-ones bearing quaternary stereocenters (Scheme 1) [14]. Notably, this work represents the first example of 1H-pyrrole-2,3-diones used as Henry acceptors for the asymmetric reaction. Herein, we report our preliminary results on this subject.
![[1860-5397-12-31-i1]](/bjoc/content/inline/1860-5397-12-31-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The strategy to construct chiral 3-substituted-3-hydroxy-1H-pyrrol-2(3H)-ones.
Scheme 1: The strategy to construct chiral 3-substituted-3-hydroxy-1H-pyrrol-2(3H)-ones.
Results and Discussion
We started our studies with the reaction of ethyl 1-benzyl-4,5-dioxo-2-phenyl-4,5-dihydro-1H-pyrrole-3-carboxylate (1a) and nitromethane (2a) in the presence of various chiral bifunctional organocatalysts 3a–e in dichloromethane (Table 1). As expected, the reaction proceeded and gave the desired product 4a in 18% yield and 28% ee with cinchonidine and L-valine-based catalyst 3a (Table 1, entry 1). The bifunctional, thiourea-tertiary amine catalyst 3b, derived from quinine and L-valine, furnished a similar result to catalyst 3a (Table 1, entry 2). Next, the reaction was attempted with catalyst 3c derived from L-phenylalanine and catalyst 3d derived from L-phenylglycine, and improvements in enantioselectivity were observed (Table 1, entries 3 and 4). Changing L-phenylalanine to D-phenylalanine furnished catalyst 3e, and the enantioselectivity was improved to 61% ee (Table 1, entry 5). Having identified 3e as the best catalyst, we undertook a solvent screening for this transformation with 20 mol % 3e at 30 °C (Table 1, entries 6–12). Arenes such as toluene and mesitylene gave relatively lower ee values (Table 1, entries 6 and 7). In contrast, the reactions in ethers gave improved yield with slightly increased enantioselectivity (Table 1, entries 8–10). However, strong polar solvents such as acetonitrile and ethyl acetate proved inferior to this Henry reaction (Table 1, entries 11 and 12). The solvent survey revealed that THF was the suitable solvent in terms of the yield and enantioselectivity (Table 1, entry 8).
Table 1: Screening of the catalysts and solvents.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-31-i2.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Solvent | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 3a | DCM | 18 | 28 |
| 2 | 3b | DCM | 25 | 27 |
| 3 | 3c | DCM | 25 | 38 |
| 4 | 3d | DCM | 29 | 41 |
| 5 | 3e | DCM | 23 | 61 |
| 6 | 3e | toluene | 24 | 46 |
| 7 | 3e | mesitylene | 36 | 58 |
| 8 | 3e | THF | 61 | 61 |
| 9 | 3e | dioxane | 59 | 53 |
| 10 | 3e | Et2O | 48 | 62 |
| 11 | 3e | CH3CN | 14 | 40 |
| 12 | 3e | EtOAc | 18 | 59 |
aUnless otherwise noted, the reactions were carried out with 1a (0.2 mmol), 2a (2.0 mmol), catalyst 3 (20 mol %) in solvent (2 mL) at 30 °C for 14 h. bIsolated yield. cDetermined by chiral HPLC analysis.
To further optimize the reaction conditions, the substituent at the nitrogen atom in 1H-pyrrole-2,3-diones 1 and the substrate concentration were investigated. The results are summarized in Table 2. It was found that an isopropyl group furnished a better yield and ee value than a benzyl group (Table 2, entry 2 vs entry 1). Changing the isopropyl group to a methyl group decreased the enantioselectivity (Table 2, entry 3). To enhance the reactivity of the substrate, we also tried to introduce some electron-withdrawing substituents on the nitrogen atom. However, we could not obtain the desired substrates. Afterwards, upon the investigation of substrate concentration (Table 2, entries 4 and 5), it was found that increasing the substrate concentration could slightly increase the yield with unchanged enantioselectivity (Table 2, entry 4). Ultimately, the effects of the reaction temperature and the additive were also examined and no improved results were obtained (Table 2, entries 6–8). Based on these observations, the most appropriate conditions for the Henry reaction could be established: 1 equiv of 1a and 10 equiv of 2a with 20 mol % catalyst 3e in 1.0 mL THF at 30 °C (Table 2, entry 4).
Table 2: Further optimization of conditions.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-31-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | 1 | x | Time (h) | 4 | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 1a | 2 | 14 | 4a | 61 | 61 |
| 2 | 1b | 2 | 14 | 4b | 64 | 71 |
| 3 | 1c | 2 | 14 | 4c | 19 | 57 |
| 4 | 1b | 1 | 14 | 4b | 69 | 71 |
| 5 | 1b | 3 | 14 | 4b | 60 | 70 |
| 6d | 1b | 1 | 14 | 4b | 64 | 46 |
| 7e | 1b | 1 | 62 | 4b | trace | ND |
| 8f | 1b | 1 | 14 | 4b | 75 | 66 |
aUnless otherwise noted, the reactions were carried out with 1a (0.2 mmol), 2a (2.0 mmol) and catalyst 3e (20 mol %) in THF for the specified reaction time. bIsolated yield. cDetermined by chiral HPLC analysis. dRun at 50 °C; eRun at 0 °C. f4 Å MS was added. ND, not determined.
Under the optimal reaction conditions, the substrate scope and the limitation of this organocatalytic Henry reaction were explored. The results are summarized in Table 3. First, we investigated the effects of the alkyl ester of 1H-pyrrole-2,3-diones 1. The replacement of the ethyl group by either a methyl or a butyl group had only a slight effect on the ee value but an obvious effect on the yield (Table 3, entries 1 and 2 vs Table 2, entry 4). After introducing different substituent groups on the phenyl ring of substrate 1, it was observed that the reactions take place, and the corresponding products in 44–73% yield with up to 73% ee were furnished. This was true regardless of the electronic nature and position of the substituents on the phenyl ring (Table 3, entries 3–10). When the substituent was in the ortho-position of the phenyl ring of the R2 group, a diastereoselectivity was observed that may be due to the steric hindrance of ortho substituents led to the atropisomers (Table 3, entry 3). A similar result was observed for the bulky 1-naphthyl group (Table 3, entry 11). The reaction was also performed with a 2-thienyl residue and the desired product was obtained in 51% yield and 71% ee (Table 3, entry 10). In addition, substrate 1o with no substituent on the nitrogen atom gave the desired product in 41% and 48% ee (Table 3, entry 12). The introduction of a phenyl group on the nitrogen atom had a negative effect on the reaction (Table 3, entry 13). Nitroethane was also used to react with 1H-pyrrole-2,3-dione 1b, and a good diastereoselectivity was achieved with an unfavorable yield and ee value (Table 3, entry 14). For the substrates with methoxy groups on the phenyl rings, the reaction gave relatively better results (Table 3, entries 4 and 7). We also tested the substrates 1q and 1r containing a methyl ester, and acceptable results were obtained (Table 3, entries 15 and 16). The absolute configuration of the major isomer 4i was unambiguously determined to be S by single-crystal X-ray analysis (Figure 1) [38]. The configurations of the other products were assigned by analogy.
Table 3: Scope of the reaction.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-31-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R/ R1/R2 | 2 | Time (h) | 4 | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | iPr/Me/Ph (1d) | 2a | 14 | 4d | 55 | 65 |
| 2 | iPr/n-Bu/Ph (1e) | 2a | 70 | 4e | 52 | 70 |
| 3 | iPr/Et/2-ClC6H4 (1f) | 2a | 62 | 4f | 53 | 73d |
| 4 | iPr/Et/3-MeOC6H4 (1g) | 2a | 62 | 4g | 73 | 71 |
| 5 | iPr/Et/3-ClC6H4 (1h) | 2a | 120 | 4h | 47 | 71 |
| 6 | iPr/Et/3-BrC6H4 (1i) | 2a | 120 | 4i | 44 | 70 |
| 7 | iPr/Et/4-MeOC6H4 (1j) | 2a | 62 | 4j | 61 | 72 |
| 8 | iPr/Et/4-MeC6H4 (1k) | 2a | 48 | 4k | 64 | 69 |
| 9 | iPr/Et/4-FC6H4 (1l) | 2a | 62 | 4l | 73 | 70 |
| 10 | iPr/Et/2-thienyl (1m) | 2a | 120 | 4m | 51 | 71 |
| 11 | iPr/Et/1-naphthyl (1n) | 2a | 120 | 4n | 75 | 51e |
| 12 | H/Me/Ph (1o) | 2a | 120 | 4o | 41 | 48 |
| 13 | Ph/Et/Ph (1p) | 2a | 120 | 4p | 42 | 29 |
| 14 | iPr/Et/Ph (1b) | 2b | 24 | 4q | 23 | 22f |
| 15 | iPr/Me/4-MeOC6H4 (1q) | 2a | 84 | 4r | 51 | 69 |
| 16 | iPr/Me/3-MeOC6H4 (1r) | 2a | 120 | 4s | 71 | 58 |
aUnless otherwise noted, the reactions were carried out with 1 (0.2 mmol), 2 (2.0 mmol). bIsolated yield. cDetermined by chiral HPLC analysis. dee for the major isomer and ee of another isomer is 60%, and 54:46 dr was observed. eee for the major isomer and ee of another isomer is 49%, and 53:47 dr was observed. fee for the major isomer, and 94:6 dr was observed.
![[1860-5397-12-31-1]](/bjoc/content/figures/1860-5397-12-31-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The X-ray structure of compound 4i.
Figure 1: The X-ray structure of compound 4i.
On the basis of our experimental results and the related reports about the bifunctional activation mode of nitromethane with different electrophiles [14,17,39], we propose a possible model to explain the stereochemistry of this transformation. As shown in Figure 2, nitromethane is activated by the tertiary amine to form the nitro enolate. Simultaneously, the 1H-pyrrole-2,3-diones 1 are orientated by the multiple hydrogen bonds of the catalyst. Thus, the nitro enolate attacks the keto carbonyl group of 1H-pyrrole-2,3-diones (to the si-face) to furnish the corresponding product with S-configuration (Figure 2).
![[1860-5397-12-31-2]](/bjoc/content/figures/1860-5397-12-31-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A proposed transition state for the asymmetric Henry reaction.
Figure 2: A proposed transition state for the asymmetric Henry reaction.
Conclusion
In conclusion, we have developed an asymmetric Henry reaction of 1H-pyrrole-2,3-diones with a chiral bifunctional amine-thiourea possessing multiple hydrogen-bond donors as the catalyst. With the developed protocol, a range of 3-hydroxy-3-nitromethyl-1H-pyrrol-2(3H)-ones bearing quaternary stereocenters were obtained in good yield (up to 75%) and with moderate to good enantioselectivity (up to 73% ee). A possible transition-state model, characterized by the bifunctional catalyst acting as a multiple hydrogen-bond donor, is also proposed. The application of 1H-pyrrole-2,3-diones in the catalytic asymmetric reactions for the preparation of biologically relevant compounds is currently underway.
References
-
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650
Return to citation in text: [1] -
Volla, C. M. R.; Atodiresei, I.; Rueping, M. Chem. Rev. 2014, 114, 2390–2431. doi:10.1021/cr400215u
Return to citation in text: [1] -
Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703–4832. doi:10.1021/cr100348t
Return to citation in text: [1] -
Wei, Y.; Shi, M. Chem. Rev. 2013, 113, 6659–6690. doi:10.1021/cr300192h
Return to citation in text: [1] -
Chauhan, P.; Mahajan, S.; Enders, D. Chem. Rev. 2014, 114, 8807–8864. doi:10.1021/cr500235v
Return to citation in text: [1] -
Zhu, Y.; Wang, Q.; Cornwall, R. G.; Shi, Y. Chem. Rev. 2014, 114, 8199–8256. doi:10.1021/cr500064w
Return to citation in text: [1] -
Connon, S. J. Chem. – Eur. J. 2006, 12, 5418–5427. doi:10.1002/chem.200501076
Return to citation in text: [1] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r
Return to citation in text: [1] -
Akiyama, T. Chem. Rev. 2007, 107, 5744–5758. doi:10.1021/cr068374j
Return to citation in text: [1] -
Palomo, C.; Oiarbide, M.; López, R. Chem. Soc. Rev. 2009, 38, 632–653. doi:10.1039/B708453F
Return to citation in text: [1] -
Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem. – Eur. J. 2011, 17, 6890–6899. doi:10.1002/chem.201003694
Return to citation in text: [1] -
Fang, X.; Wang, C.-J. Chem. Commun. 2015, 51, 1185–1197. doi:10.1039/C4CC07909D
Return to citation in text: [1] -
Wang, C.-J.; Dong, X.-Q.; Zhang, Z.-H.; Xue, Z.-Y.; Teng, H.-L. J. Am. Chem. Soc. 2008, 130, 8606–8607. doi:10.1021/ja803538x
Return to citation in text: [1] -
Zhu, Q.; Lu, Y. Angew. Chem., Int. Ed. 2010, 49, 7753–7756. doi:10.1002/anie.201003837
Return to citation in text: [1] [2] [3] -
Palomo, C.; Oiarbide, M.; Laso, A. Eur. J. Org. Chem. 2007, 2561–2574. doi:10.1002/ejoc.200700021
Return to citation in text: [1] -
Alvarez-Casao, Y.; Marques-Lopez, E.; Herrera, R. P. Symmetry 2011, 3, 220–245. doi:10.3390/sym3020220
Return to citation in text: [1] -
Marcelli, T.; van der Haas, R. N. S.; van Maarseveen, J. H.; Hiemstra, H. Angew. Chem., Int. Ed. 2006, 45, 929–931. doi:10.1002/anie.200503724
Return to citation in text: [1] [2] -
Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Adv. Synth. Catal. 2005, 347, 1643–1648. doi:10.1002/adsc.200505148
Return to citation in text: [1] -
Sohtome, Y.; Takemura, N.; Takada, K.; Takagi, R.; Iguchi, T.; Nagasawa, K. Chem. – Asian J. 2007, 2, 1150–1160. doi:10.1002/asia.200700145
Return to citation in text: [1] -
Li, H.; Wang, B.; Deng, L. J. Am. Chem. Soc. 2006, 128, 732–733. doi:10.1021/ja057237l
Return to citation in text: [1] -
Mandal, T.; Samanta, S.; Zhao, C.-G. Org. Lett. 2007, 9, 943–945. doi:10.1021/ol070209i
Return to citation in text: [1] -
Chen, X.; Wang, J.; Zhu, Y.; Shang, D.; Gao, B.; Liu, X.; Feng, X.; Su, Z.; Hu, C. Chem. – Eur. J. 2008, 14, 10896–10899. doi:10.1002/chem.200801958
Return to citation in text: [1] -
Bandini, M.; Sinisi, R.; Umani-Ronchi, A. Chem. Commun. 2008, 4360–4362. doi:10.1039/b807640e
Return to citation in text: [1] -
Vlatković, M.; Bernardi, L.; Otten, E.; Feringa, B. L. Chem. Commun. 2014, 50, 7773–7775. doi:10.1039/c4cc00794h
Return to citation in text: [1] -
Liu, L.; Zhang, S.; Xue, F.; Lou, G.; Zhang, H.; Ma, S.; Duan, W.; Wang, W. Chem. – Eur. J. 2011, 17, 7791–7795. doi:10.1002/chem.201101025
Return to citation in text: [1] -
Zhang, Y.; Li, Z. J.; Xu, H. S.; Zhang, Y.; Wang, W. RSC Adv. 2011, 1, 389–392. doi:10.1039/c1ra00477h
Return to citation in text: [1] -
Fürstner, A. Angew. Chem., Int. Ed. 2003, 42, 3582–3603. doi:10.1002/anie.200300582
Return to citation in text: [1] -
Larionov, O. V.; de Meijere, A. Angew. Chem., Int. Ed. 2005, 44, 5664–5667. doi:10.1002/anie.200502140
Return to citation in text: [1] -
Mylari, B. L.; Beyer, T. A.; Siegel, T. W. J. Med. Chem. 1991, 34, 1011–1018. doi:10.1021/jm00107a020
Return to citation in text: [1] -
Silaichev, P. S.; Dmitriev, M. V.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2010, 46, 1173–1176. doi:10.1134/S1070428010080105
Return to citation in text: [1] -
Dmitriev, M. V.; Silaichev, P. S.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 1165–1168. doi:10.1134/S1070428011080082
Return to citation in text: [1] -
Dmitriev, M. V.; Silaichev, P. S.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 304–305. doi:10.1134/S1070428011020266
Return to citation in text: [1] -
Bhanushali, M.; Zhao, C.-G. Tetrahedron Lett. 2012, 53, 359–362. doi:10.1016/j.tetlet.2011.11.056
Return to citation in text: [1] -
Zhang, M.-L.; Chen, L.; You, Y.; Wang, Z.-H.; Yue, D.-F.; Zhang, X.-M.; Xu, X.-Y.; Yuan, W.-C. Tetrahedron 2016, in press. doi:10.1016/j.tet.2015.08.046
Return to citation in text: [1] -
Zhao, J.-Q.; Zhou, M.-Q.; Wu, Z.-J.; Wang, Z.-H.; Yue, D.-F.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2015, 17, 2238–2241. doi:10.1021/acs.orglett.5b00850
Return to citation in text: [1] -
Wang, Z.-H.; Wu, Z.-J.; Huang, X.-Q.; Yue, D.-F.; You, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Chem. Commun. 2015, 51, 15835–15838. doi:10.1039/C5CC06383C
Return to citation in text: [1] -
Liu, X.-L.; Han, W.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2013, 15, 1246–1249. doi:10.1021/ol400183k
Return to citation in text: [1] -
See Supporting Information File 2 for crystallographic data. The CCDC number is 1435541. The level of probability of the thermal ellipsoids is 35%.
Return to citation in text: [1] -
Li, M.-Q.; Zhang, J.-X.; Huang, X.-F.; Wu, B.; Liu, Z.-M.; Chen, J.; Li, X.-D.; Wang, X.-W. Eur. J. Org. Chem. 2011, 5237–5241. doi:10.1002/ejoc.201100824
Return to citation in text: [1]
| 14. | Zhu, Q.; Lu, Y. Angew. Chem., Int. Ed. 2010, 49, 7753–7756. doi:10.1002/anie.201003837 |
| 17. | Marcelli, T.; van der Haas, R. N. S.; van Maarseveen, J. H.; Hiemstra, H. Angew. Chem., Int. Ed. 2006, 45, 929–931. doi:10.1002/anie.200503724 |
| 39. | Li, M.-Q.; Zhang, J.-X.; Huang, X.-F.; Wu, B.; Liu, Z.-M.; Chen, J.; Li, X.-D.; Wang, X.-W. Eur. J. Org. Chem. 2011, 5237–5241. doi:10.1002/ejoc.201100824 |
| 14. | Zhu, Q.; Lu, Y. Angew. Chem., Int. Ed. 2010, 49, 7753–7756. doi:10.1002/anie.201003837 |
| 38. | See Supporting Information File 2 for crystallographic data. The CCDC number is 1435541. The level of probability of the thermal ellipsoids is 35%. |
| 1. | Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650 |
| 2. | Volla, C. M. R.; Atodiresei, I.; Rueping, M. Chem. Rev. 2014, 114, 2390–2431. doi:10.1021/cr400215u |
| 3. | Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703–4832. doi:10.1021/cr100348t |
| 4. | Wei, Y.; Shi, M. Chem. Rev. 2013, 113, 6659–6690. doi:10.1021/cr300192h |
| 5. | Chauhan, P.; Mahajan, S.; Enders, D. Chem. Rev. 2014, 114, 8807–8864. doi:10.1021/cr500235v |
| 6. | Zhu, Y.; Wang, Q.; Cornwall, R. G.; Shi, Y. Chem. Rev. 2014, 114, 8199–8256. doi:10.1021/cr500064w |
| 16. | Alvarez-Casao, Y.; Marques-Lopez, E.; Herrera, R. P. Symmetry 2011, 3, 220–245. doi:10.3390/sym3020220 |
| 34. | Zhang, M.-L.; Chen, L.; You, Y.; Wang, Z.-H.; Yue, D.-F.; Zhang, X.-M.; Xu, X.-Y.; Yuan, W.-C. Tetrahedron 2016, in press. doi:10.1016/j.tet.2015.08.046 |
| 15. | Palomo, C.; Oiarbide, M.; Laso, A. Eur. J. Org. Chem. 2007, 2561–2574. doi:10.1002/ejoc.200700021 |
| 35. | Zhao, J.-Q.; Zhou, M.-Q.; Wu, Z.-J.; Wang, Z.-H.; Yue, D.-F.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2015, 17, 2238–2241. doi:10.1021/acs.orglett.5b00850 |
| 36. | Wang, Z.-H.; Wu, Z.-J.; Huang, X.-Q.; Yue, D.-F.; You, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Chem. Commun. 2015, 51, 15835–15838. doi:10.1039/C5CC06383C |
| 37. | Liu, X.-L.; Han, W.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2013, 15, 1246–1249. doi:10.1021/ol400183k |
| 12. | Fang, X.; Wang, C.-J. Chem. Commun. 2015, 51, 1185–1197. doi:10.1039/C4CC07909D |
| 13. | Wang, C.-J.; Dong, X.-Q.; Zhang, Z.-H.; Xue, Z.-Y.; Teng, H.-L. J. Am. Chem. Soc. 2008, 130, 8606–8607. doi:10.1021/ja803538x |
| 14. | Zhu, Q.; Lu, Y. Angew. Chem., Int. Ed. 2010, 49, 7753–7756. doi:10.1002/anie.201003837 |
| 30. | Silaichev, P. S.; Dmitriev, M. V.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2010, 46, 1173–1176. doi:10.1134/S1070428010080105 |
| 31. | Dmitriev, M. V.; Silaichev, P. S.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 1165–1168. doi:10.1134/S1070428011080082 |
| 32. | Dmitriev, M. V.; Silaichev, P. S.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 304–305. doi:10.1134/S1070428011020266 |
| 7. | Connon, S. J. Chem. – Eur. J. 2006, 12, 5418–5427. doi:10.1002/chem.200501076 |
| 8. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 9. | Akiyama, T. Chem. Rev. 2007, 107, 5744–5758. doi:10.1021/cr068374j |
| 10. | Palomo, C.; Oiarbide, M.; López, R. Chem. Soc. Rev. 2009, 38, 632–653. doi:10.1039/B708453F |
| 11. | Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem. – Eur. J. 2011, 17, 6890–6899. doi:10.1002/chem.201003694 |
| 33. | Bhanushali, M.; Zhao, C.-G. Tetrahedron Lett. 2012, 53, 359–362. doi:10.1016/j.tetlet.2011.11.056 |
| 23. | Bandini, M.; Sinisi, R.; Umani-Ronchi, A. Chem. Commun. 2008, 4360–4362. doi:10.1039/b807640e |
| 24. | Vlatković, M.; Bernardi, L.; Otten, E.; Feringa, B. L. Chem. Commun. 2014, 50, 7773–7775. doi:10.1039/c4cc00794h |
| 27. | Fürstner, A. Angew. Chem., Int. Ed. 2003, 42, 3582–3603. doi:10.1002/anie.200300582 |
| 28. | Larionov, O. V.; de Meijere, A. Angew. Chem., Int. Ed. 2005, 44, 5664–5667. doi:10.1002/anie.200502140 |
| 21. | Mandal, T.; Samanta, S.; Zhao, C.-G. Org. Lett. 2007, 9, 943–945. doi:10.1021/ol070209i |
| 22. | Chen, X.; Wang, J.; Zhu, Y.; Shang, D.; Gao, B.; Liu, X.; Feng, X.; Su, Z.; Hu, C. Chem. – Eur. J. 2008, 14, 10896–10899. doi:10.1002/chem.200801958 |
| 29. | Mylari, B. L.; Beyer, T. A.; Siegel, T. W. J. Med. Chem. 1991, 34, 1011–1018. doi:10.1021/jm00107a020 |
| 20. | Li, H.; Wang, B.; Deng, L. J. Am. Chem. Soc. 2006, 128, 732–733. doi:10.1021/ja057237l |
| 17. | Marcelli, T.; van der Haas, R. N. S.; van Maarseveen, J. H.; Hiemstra, H. Angew. Chem., Int. Ed. 2006, 45, 929–931. doi:10.1002/anie.200503724 |
| 18. | Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Adv. Synth. Catal. 2005, 347, 1643–1648. doi:10.1002/adsc.200505148 |
| 19. | Sohtome, Y.; Takemura, N.; Takada, K.; Takagi, R.; Iguchi, T.; Nagasawa, K. Chem. – Asian J. 2007, 2, 1150–1160. doi:10.1002/asia.200700145 |
| 25. | Liu, L.; Zhang, S.; Xue, F.; Lou, G.; Zhang, H.; Ma, S.; Duan, W.; Wang, W. Chem. – Eur. J. 2011, 17, 7791–7795. doi:10.1002/chem.201101025 |
| 26. | Zhang, Y.; Li, Z. J.; Xu, H. S.; Zhang, Y.; Wang, W. RSC Adv. 2011, 1, 389–392. doi:10.1039/c1ra00477h |
© 2016 Zhang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)